DOI:
10.1039/C5RA21264B
(Paper)
RSC Adv., 2015,
5, 99632-99639
Facile synthesis of nanostructured LiMnPO4 as a high-performance cathode material with long cycle life and superior rate capability†
Received
13th October 2015
, Accepted 6th November 2015
First published on 10th November 2015
Abstract
Lithium manganese phosphate (LiMnPO4) has been considered as an alternative to lithium iron phosphate (LiFePO4) for next-generation Li-ion battery cathodes because of its higher working voltage. However, facile preparation methods for high-performance LiMnPO4 are still lacking. In this work, we propose a facile route to prepare nano-LiMnPO4 (30–50 nm) by using citric acid (CA) as a surfactant. The addition of a small amount of CA in the precursor leads to an obvious size reduction of LiMnPO4. After carbon-coating, nano-LiMnPO4 exhibits excellent rate capability and long cycle life at high rates because of the small size and uniform/thin carbon coating. At a high rate of up to 20C (3.4 A g−1), LiMnPO4/C can still deliver a high discharge capacity of 96.6 mA h g−1. LiMnPO4/C also exhibits long cycle life with ∼70% capacity retained after 500 cycles at 10C. The excellent electrochemical performance of LiMnPO4/C makes it an attractive cathode in high-power and high-energy Li-ion batteries.
1. Introduction
LiMPO4 (M = Fe, Mn, Co) with an olivine-type structure has gained wide interest as a new cathode material for Li-ion batteries since the first report on LiFePO4 by Goodenough and co-workers in 1997.1 In these olivine-type materials, LiFePO4 has now realized practical applications in electric vehicles (EVs) because of its environmental friendliness, low cost and structural stability.2,3 Compared with LiFePO4, LiMnPO4 (LMP) could provide a larger energy density with its higher redox potential of Mn2+/Mn3+ (4.1 V vs. Li/Li+) compared to Fe2+/Fe3+ (3.45 V vs. Li/Li+).4–6 However, LiMnPO4 exhibits a rather lower electrochemical activity than LiFePO4 due to its intrinsically lower electronic and ionic conductivity,7,8 the structural instability of the MnPO4 phase,9,10 and the larger volume change between LiMnPO4 and MnPO4.11 In addition, Mn3+ in the charged state undergoes Jahn–Teller distortion.12,13 In recent years, a great effort has been made to improve the electrochemical activity of LiMnPO4 through cation substitution, size decrease, optimized carbon coating, etc.14 Cation substitution has been found to be an effective measure to activate LiMnPO4 and stabilize the delithiated phase.15 However, the substitution should be controlled at a low level to maintain the high energy density of LiMnPO4.16–24
Size decrease is another useful method to enhance the electrochemical activity of LiMnPO4. Oh et al. synthesized LiMnPO4 using a spray-pyrolysis/ball-milling route.25 LiMnPO4 of 10–50 nm could deliver high capacities of 158 mA h g−1 at 0.05C and 126 mA h g−1 at 1C after coating with a uniform carbon layer. Recent work has shown that nano-engineering could remarkably improve the electrochemical performance of LiMnPO4.18,26–42 Since Yang et al. first reported the direct synthesis of LiFePO4 using a hydrothermal method,43 hydrothermal/solvothermal routes have been widely used to prepare LiMPO4 (M = Fe, Mn) with a nanostructure.44 The size and morphology of LiMnPO4 can be easily regulated by controlling the synthetic conditions (temperature, time, reactant concentration/ratio, etc.) and using different solvents or surfactants.26–29,35–39 The work by Qin et al. indicated that the morphology of LiMnPO4 can be controlled by simply adjusting the pH value.27 The obtained LiMnPO4 nanoplates could yield high capacities of 149 mA h g−1 at 0.1C and 90 mA h g−1 at 1C after graphene coating. Hong et al. synthesized LiMnPO4 nanorods by setting the volume ratio of ethylene glycol (EG) and water at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.37 The carbon-coated LiMnPO4 could deliver a high capacity of 110 mA h g−1 at 10C and a capacity retention of ∼94.5% after 100 cycles at 0.5C.
:1.37 The carbon-coated LiMnPO4 could deliver a high capacity of 110 mA h g−1 at 10C and a capacity retention of ∼94.5% after 100 cycles at 0.5C.
It is generally accepted that nano-engineering is a practical strategy to realize the high performance of LiMnPO4 materials. Nevertheless, a challenge still remains to find a facile preparation method for nanosized LiMnPO4. For the solvothermal synthesis of LiMnPO4, the reaction of H3PO4 + 3LiOH + MnSO4 = LiMnPO4 + Li2SO4 + 3H2O is usually adopted. The morphology of LiMnPO4 was found to depend greatly on the molar ratios of H3PO4/LiOH/MnSO4.27,39 Actually, acidity plays a critical role in determining the morphology of LiMnPO4 in the reactions. In this work, nanostructured LiMnPO4 was prepared using a facile solvothermal route in EG/H2O mixed solvent with citric acid (CA) as a surfactant. The results showed that the addition of a small amount of CA leads to an obvious size decrease and a considerable performance improvement of LiMnPO4. The LiMnPO4/C granules of 30–50 nm could deliver high capacities of 147.9, 113.0 and 96.6 mA h g−1 at 1C, 10C and 20C, respectively. The capacities can be retained at 89.1 and 80.5 mA h g−1 after 500 cycles at 1C and 10C, respectively. The intrinsic mechanism for performance enhancement was also investigated using cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS). This work provides a facile method to realize high performance of LiMnPO4 materials.
2. Experimental section
2.1 Preparation of LiMnPO4 and LiMnPO4/C
LiMnPO4 was prepared using a facile solvothermal route via the reaction H3PO4 + 3LiOH + MnSO4 = LiMnPO4 + Li2SO4 + 3H2O in EG/water mixed solvent.39 The molar ratio of H3PO4 to LiOH to MnSO4 in the precursor is 1:3:1. During the preparation of the MnSO4 solution in EG/water, the desired amount of CA (1–7 mmol) was added. The reaction products are named LMP-x, where x represents the amount of CA used (in the unit of mmol). For example, when 1.0 mmol of CA was used, the product is named LMP-1.0. The carbon coating procedure was conducted according to previous work.39 For simplicity, LiMnPO4/C uses the same name as the corresponding LiMnPO4 sample.
2.2 Material characterization
X-ray diffraction (XRD) patterns were collected on a Rigaku D/Max-2550pc powder diffractometer (Cu Kα, λ = 0.1541 nm) to analyze the crystalline structure of LiMnPO4. The morphology and microstructure of LiMnPO4 and LiMnPO4/C were checked using scanning electron microscopy (SEM) on an S-4800 microscope and transmission electron microscopy (TEM) on a JEM 2100F microscope. The carbon content in the LiMnPO4/C samples was measured using a Flash EA 1112 tester. In this equipment, the solid carbon can be combusted into gaseous CO2 in a rapid and dynamic mode. The carbon content can be determined by analyzing the amount of CO2 with chromatography. The Brunauer–Emmett–Teller (BET) specific surface area of LiMnPO4 was calculated based on the N2 absorption/desorption isotherms using a Quantachrome Autosorb-1 analyser.
2.3 Electrochemical measurements
The electrochemical performance of LiMnPO4/C was measured using CR2025-type coin cells on a Neware battery cycler (Shenzhen, China). The electrode was made by homogeneously mixing LiMnPO4/C, acetylene black and polyvinylidene fluoride in a mass ratio of 7:2:1. The active material (LiMnPO4/C) loading was around 2 mg. The cell assembly was conducted in a glove box filled with pure Ar. For the cells, metallic Li foils were used as the counter electrodes, 1 M LiPF6 in ethylene carbonate (EC)/dimethyl carbonate (DMC) (1:1 by volume) was used as the electrolyte, and Celgard 2300 microporous membranes were used as the separators. The cells were tested with a constant-current–constant-voltage (CC–CV) protocol. The cells were first galvanostatically charged to 4.5 V at different current rates, then held at 4.5 V for 1 h, and galvanostatically discharged to 2.0 V. The charge and discharge processes use the same current rate. The current density was calculated based on the total mass of LiMnPO4 and carbon for the LiMnPO4/C composites. The specific capacity of LiMnPO4/C is normalized by the mass of LiMnPO4 and 1C is defined as 170 mA g−1. CV tests were conducted at 2.0–4.5 V (vs. Li/Li+) with a scan rate of 0.1 mV s−1 on a VersaSTAT3 electrochemistry workstation (Princeton Applied Research). EIS tests were carried out on the VersaSTAT3 workstation using an ac voltage of 10 mV amplitude in a frequency range of 10 mHz to 100 kHz. The EIS measurements were performed in the lithiated state of LiMnPO4 after the cells were rested for several hours. The electrochemical tests were performed at room temperature.
3. Results and discussion
Fig. 1 gives the XRD patterns of the solvothermal products with different amounts of CA in the precursors. The phase purity of the LiMnPO4 samples was confirmed by comparing with the standard diffraction peaks of LiMnPO4 (space group pnmb, JCPDS card no. 33-0804). Note that the relative intensity of the diffraction peaks changes with an increase in the CA amount, suggesting changes in the morphology and size of LiMnPO4. The sharp peaks suggest high crystallinity in the LiMnPO4 samples even though they were prepared at low temperature.
 |
| | Fig. 1 XRD patterns of LiMnPO4 prepared with different CA amounts in the precursors. | |
The morphology of the solvothermal products was characterized using SEM. The LiMnPO4 exhibits a spindle-like shape when it was prepared with a CA-free precursor (Fig. S1†). The size of the spindle-like LiMnPO4 is around 200 nm and the BET specific surface area is 32.2 m2 g−1. As seen in Fig. 2a, the shape and size of LiMnPO4 show a remarkable change when a small amount of CA (CA/MnSO4 molar ratio is 1/10) was added in the precursor. The obtained LiMnPO4 demonstrates a plate-like shape with a size below 100 nm. The size of LiMnPO4 can be further reduced to 30–50 nm by increasing the CA amount (CA/MnSO4 molar ratio is 3/10). The LiMnPO4 exhibits an irregular granule shape with the BET surface area increasing to 53.8 m2 g−1 (Fig. 2b). The size of LiMnPO4 shows a trend to increase when the amount of CA was further increased (Fig. 2c–f). At a CA/MnSO4 molar ratio of 7/10, the LiMnPO4 crystallizes into a plate-like shape again and the surface area decreases to 45.7 m2 g−1 (Fig. 2f). Even so, the plate-like LiMnPO4 still has a smaller size than the spindle-like one, suggesting that CA does play a critical role in reducing the size of LiMnPO4. This size decrease, in turn, enhances the electrochemical performance of LiMnPO4, which will be discussed later.
 |
| | Fig. 2 SEM images of LiMnPO4 prepared with different CA/MnSO4 molar ratios in the precursors: (a) 1/10, (b) 3/10, (c) 7/20, (d) 2/5, (e) 1/2 and (f) 7/10. | |
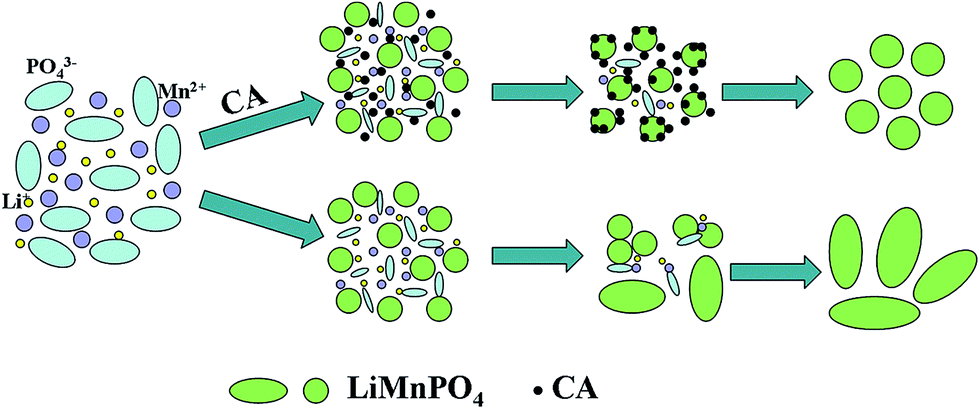
Fig. 3 presents TEM images of the pristine LMP-3.0 and carbon-coated LMP-3.0. As seen in Fig. 3b, the pristine LMP-3.0 exhibits an irregular shape with a size of 30–50 nm, agreeing with the SEM observations. The morphology of the sample was retained after carbon coating as shown in Fig. 3c. The high-resolution TEM (HRTEM) image in Fig. 3d indicates that the LiMnPO4 is well crystallized. The lattice spacings of 0.21 and 0.36 nm are related to the (112) and (111) planes of LiMnPO4. The surface of LiMnPO4 is uniformly coated by a layer of carbon with a thickness of ∼1 nm. As a result, size decrease of LiMnPO4 has been realized through a facile solvothermal route using CA as the surfactant. The mechanism is schematically illustrated in Fig. 4. The adsorption of CA on the surface of LiMnPO4 inhibits its continuous growth during the solvothermal reaction. The LiMnPO4 grows into an irregular shape due possibly to the different adsorption ability of CA on the different crystalline planes of LiMnPO4. However, excess CA will greatly change the acidity of the solution, leading to the formation of plate-like LiMnPO4, similar to the case of H3PO4 where excess H3PO4 also results in the crystal growth of LiMnPO4.39
 |
| | Fig. 3 TEM images of (a, b) pristine and (c, d) carbon-coated LMP-3.0. | |
 |
| | Fig. 4 Schematic illustration of the CA-induced restrained growth of LiMnPO4 crystals. | |
Electrochemical tests were performed on three LiMnPO4/C samples with different sizes to reveal the size dependence of the electrochemical performance. Fig. 5a gives the first charge–discharge curves of the LiMnPO4/C samples at 0.05C. The capacities of LiMnPO4/C were calculated normalized to the mass of LiMnPO4. As seen in the figure, these samples exhibit high electrochemical activity at a low current rate, delivering high discharge capacities (164.5 mA h g−1 for LMP-3.0, 163 mA h g−1 for LMP-3.5, 161 mA h g−1 for LMP-4.0). Specially, LMP-3.0 yields the highest discharge capacity of 164.5 mA h g−1, which is close to the theoretical capacity of LiMnPO4 (170 mA h g−1). The highest capacity of LMP-3.0 is closely correlated with it having the smallest crystal size which maximizes the utilization of active material. For LiMnPO4 materials, irreversible capacities in the first cycle are usually observed, which is attributed to the passivation of the electrolyte and electrode at high potentials.37,45 The irreversible capacities of LMP-3.0, LMP-3.5 and LMP-4.0 are 17.5, 19 and 24 mA h g−1, respectively. Fig. 5b shows the CV scans of the samples at 0.1 mV s−1. LMP-3.0 displays obviously stronger and sharper current peaks than LMP-3.5 and LMP-4.0, indicating that it has the fastest electrochemical reaction kinetics due to its crystal size being the smallest.
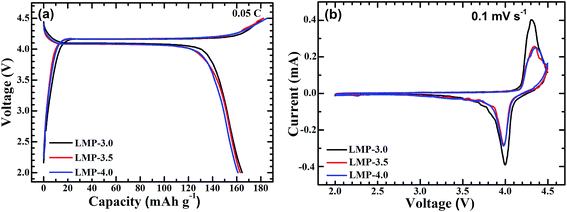 |
| | Fig. 5 (a) Voltage profiles and (b) CV plots of carbon-coated LiMnPO4. | |
Fig. 6 compares the rate capability of the LiMnPO4/C samples at current rates of 0.1–20C. The charge and discharge processes of the cells were performed at the same current rates in the rate capability tests. Note that the plateau length decreases with an increase in the current rate. The polarization also increases with an increase in the current density. LMP-3.0 shows the best rate capability among the three samples. The discharge capacities of LMP-3.0 are 158.6, 152.3, 147.9, 140.0 and 126.1 mA h g−1 at 0.1C, 0.5C, 1C, 2C and 5C, respectively. At 10C and 20C, this sample can still deliver high capacities of 113.0 and 96.6 mA h g−1, respectively. The superior rate capability of LMP-3.0 can be ascribed to its small crystal size and uniform/thin conductive carbon layer, making rapid electron and Li-ion transport possible. The LMP-3.0 sample shows a slower capacity decrease with current density than LMP-3.5 and LMP-4.0 especially at high current densities, implying that the crystal size does exert an obvious effect on Li-ion transport at the electrode/electrolyte interface and in bulk crystals.
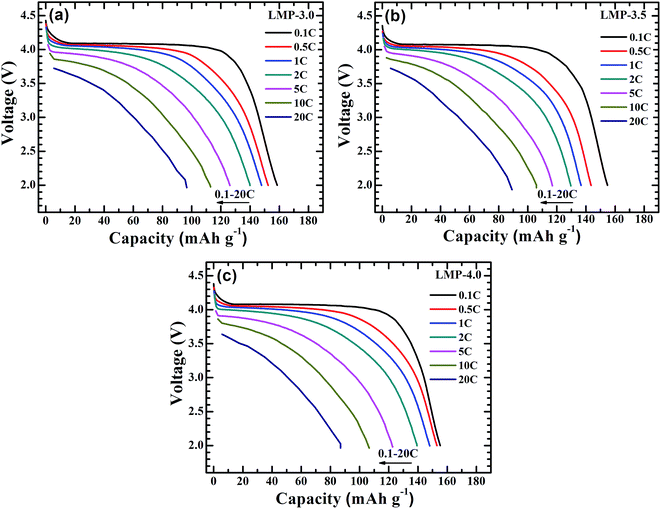 |
| | Fig. 6 Rate capability of LiMnPO4/C: (a) LMP-3.0, (b) LMP-3.5 and (c) LMP-4.0. | |
Fig. 7 demonstrates the cycling stability of the LiMnPO4/C samples. As seen in Fig. 7a, LMP-3.0, LMP-3.5 and LMP-4.0 can deliver high initial discharge capacities of 147.2, 142.8 and 142.6 mA h g−1 at 1C, which can be maintained at 89.1, 84.3 and 75.1 mA h g−1 after 500 cycles. LMP-3.0 exhibits the best cycling stability with a capacity retention of over 60% after 500 cycles at 1C. Even after 500 cycles at 10C, this sample can still keep a discharge capacity of 80.5 mA h g−1, with a retention of around 70%. Although the cycling stability of LiMnPO4/C has been enhanced by various strategies in recent work,25–30,32,33,35–37,41,42 there are few reports on LiMnPO4/C that can sustain 500 cycles at such a high current density (10C). It should be stressed that the charge and discharge in this work were performed at the same current rate. The outstanding cycling stability of LMP-3.0 could be due to the uniform carbon coating which prevents Mn dissolution,25,34,42,45 and the small crystal size which alleviates the volume strain between LiMnPO4 and MnPO4.11,42,46,47 In contrast, the large-sized spindle-like LiMnPO4 exhibits low capacity and poor cycling stability (Fig. S2†). The low capacity and poor cycling stability of spindle-like LiMnPO4 can be attributed to a low Li-ion diffusion rate with the insufficient utilization of active material and poor carbon coating for large-sized LiMnPO4 particles. Table 1 compares the rate capability and cycle life of some LiMnPO4/C composites from this work and others. The data summarized in Table 1 represent the best data on LiMnPO4/C materials reported to date. Of note is that both the rate capability and cycle life of our LMP-3.0 sample are among the best when we compare the charge/discharge mode, applied current rate and cycle number comprehensively. We propose that the outstanding electrochemical properties of our LiMnPO4/C can be attributed to the small size and uniform carbon coating, which lead to rapid electron and Li-ion transport and the easy release of the lattice strain upon repeated cycling. Carbon coating also led to remarkably improved electrochemical performance in other cathode materials such as LiNi0.8Co0.1Mn0.1O2 and LiNi0.5Mn1.5O4 by stabilizing the structure and supplying conducting channels.48,49 In addition, the ultrathin carbon layer facilitates Li-ion diffusion across the electrode/electrolyte interface with enhanced electrode kinetics. It should be noted that the LiMnPO4/C in our work was prepared using a facile solvothermal route using small amounts of inexpensive and nontoxic citric acid.
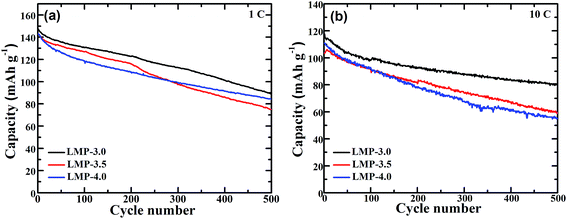 |
| | Fig. 7 Cycling stability of LiMnPO4/C at (a) 1C and (b) 10C. | |
Table 1 Comparison of the electrochemical performance of LiMnPO4/C in this work with othersa
| Sample and preparation method |
Cycling stability |
Rate capability |
Reference |
| Current density |
Initial capacity (mA h g−1) |
Cycle number |
Capacity retention |
Current density |
| Capacity (mA h g−1) |
| Note: SR = solvothermal reaction, SSR = solid state reaction, BM = ball milling, HT = high temperature, HP = high pressure, ch = charge, dis = discharge, LMP = LiMnPO4, SDBS = sodium dodecyl benzene sulfonate, G = graphene, CTAB = hexadecyltrimethyl ammonium bromide, PMMA = polymethyl methacrylate, PVP = polyvinylpyrrolidone, and OA = oleic acid. |
| LMP-3.0, SR with CA |
10C-ch, 10C-dis |
117.6 |
100/500 |
83%/68% |
1C-ch, 1C-dis |
5C-ch, 5C-dis |
10C-ch, 10C-dis |
20C-ch, 20C-dis |
This work |
| 147.9 |
126.1 |
113.0 |
96.6 |
| LMP, spray pyrolysis and BM |
0.05C-ch, 0.5C-dis |
∼140 |
50 |
94.2% |
0.05C-ch, 1C-dis |
0.05C-ch, 2C-dis |
0.05C-ch, 10C-dis |
25 |
| 126 |
107 |
∼60 |
| LMP plates, SR with SDBS |
0.05C-ch, 0.05C-dis |
147 |
50 |
93% |
0.1C-ch, 1C-dis |
0.1C-ch, 2C-dis |
0.1C-ch, 5C-dis |
26 |
| ∼110 |
∼95 |
∼70 |
| LMP/G, SR + spray drying |
1C-ch-dis, 2C-ch-dis, 5C-ch-dis |
∼90 |
60 |
75% |
1C-ch, 1C-dis |
2C-ch, 2C-dis |
5C-ch, 5C-dis |
27 |
| 90 |
∼75 |
64 |
| LMP grains, SR with CTAB |
0.05C-ch, 0.05C-dis |
153 |
110 |
95.4% |
0.05C-ch, 1C-dis |
0.05C-ch, 5C-dis |
0.05C-ch, 10C-dis |
28 |
| 128 |
111 |
92 |
| LMP, precipitation + BM |
0.05C-ch, 0.2C-dis |
∼135 |
45 |
90.5% |
0.05C-ch, 1C-dis |
0.05C-ch, 5C-dis |
0.05C-ch, 10C-dis |
30 |
| 120 |
90 |
61 |
| Porous LMP, PMMA template |
— |
0.1C-ch, 1C-dis |
0.1C-ch, 6C-dis |
0.1C-ch, 10C-dis |
31 |
| 154 |
129 |
110 |
| LMP sheets, SR with HT, HP and PVP |
0.2C-ch, 0.2C-dis |
157 |
50 |
93.6% |
5C-ch, 5C-dis |
10C-ch, 10C-dis |
20C-ch, 20C-dis |
32 |
| 119 |
93 |
63 |
| LMP granules, SSR with OA |
0.1C-ch-dis, 0.2C-ch/0.5C-dis |
122 |
50 + 50 |
97.5% + 96.4% |
0.05C-ch, 5C-dis |
0.05C-ch, 10C-dis |
0.05C-ch, 20C-dis |
33 |
| 95.7 |
87.1 |
60.1 |
| LMP plates, SR with CTAB |
0.2C-ch, 1C-dis |
130.3 |
500 |
92.7% |
1C-dis |
5C-dis |
10C-dis |
35 |
| 127.6 |
93.8 |
69.2 |
| LMP, SR |
0.5C-ch, 0.5C-dis |
138 |
100 |
91.5% |
1C-ch, 1C-dis |
5C-ch, 5C-dis |
10C-ch, 10C-dis |
36 |
| ∼135 |
118 |
106 |
| LMP rods, SR |
0.5C-ch, 0.5C-dis |
144.5 |
100 |
94.5% |
1C-ch, 1C-dis |
5C-ch, 5C-dis |
10C-ch, 10C-dis |
37 |
| 137 |
∼125 |
110 |
| LMP flakes, SR + sintering |
0.5C-ch, 0.5C-dis |
∼135 |
200 |
>95% |
1C-ch, 1C-dis |
5C-ch, 5C-dis |
10C-ch, 10C-dis |
40 |
| 130 |
110 |
92 |
| LMP, BM + SSR |
1C-ch, 1C-dis |
128 |
200 |
94% |
0.1C-ch, 1C-dis |
0.1C-ch, 2C-dis |
0.1C-ch, 5C-dis |
42 |
| >120 |
∼105 |
∼60 |
EIS tests were used to reveal the different electrode kinetics among the three samples. As seen in Fig. 8a, the Nyquist plots of the LiMnPO4/C samples are constructed with a high-frequency semicircle and a low-frequency sloping line. The plots were fitted using an equivalent circuit (see inset in Fig. 8a). In the circuit, Re denotes the electrolyte and ohmic resistance, Ri and Q1 are related to the contact resistance of the active material with the current collector and the related capacitance, respectively, Rct and Q2 represent the charge transfer resistance and double-layer capacitance, respectively, and Zw is the Warburg impedance related to Li-ion bulk diffusion.50–52 As shown in Table 2, LMP-3.0 exhibits a much lower Rct value compared with LMP-3.5 and LMP 4.0 although they have similar Ri values. The low Rct value means that there are rapid electrochemical reaction kinetics on the electrode/electrolyte interface, which is closely related to the uniform/thin conductive carbon layer and large specific surface area of LMP-3.0.
 |
| | Fig. 8 (a) Nyquist plots and equivalent circuit of LiMnPO4/C and (b) Z′ (or −Z′′) vs. ω−1/2 plots and the linear fitting of carbon-coated LMP-3.0 in the Warburg region. | |
Table 2 Fitting results of the Nyquist plots using the equivalent circuit and the DLi values
| Sample |
Re (Ω) |
Ri (Ω) |
Q1 |
Rct (Ω) |
Q2 |
DLi (cm2 s−1) |
| Y |
n |
Y |
n |
| LMP-3.0 |
3.2 |
88.5 |
9.3 × 10−5 |
0.58 |
25.4 |
1.6 × 10−5 |
0.77 |
5.0 × 10−15 |
| LMP-3.5 |
2.2 |
84.8 |
6.5 × 10−4 |
0.54 |
93.4 |
2.2 × 10−5 |
0.68 |
1.6 × 10−15 |
| LMP-4.0 |
2.7 |
84.7 |
5.9 × 10−4 |
0.58 |
117.3 |
2.1 × 10−5 |
0.67 |
2.9 × 10−15 |
Li-ion chemical diffusion coefficients DLi were also measured using EIS to further understand the different electrochemical behaviors between these LiMnPO4/C samples. To calculate the DLi values using the EIS technique, the Warburg factor σ in the Warburg region should first be determined. Fig. 8b shows the Nyquist plot of LMP-3.0 with marked frequencies f and the Warburg region with a slope of ∼45°. The inset in Fig. 8b correlates Z′ (or −Z′′) with ω−1/2 (ω = 2πf) where σ can be obtained by linearly fitting the Z′ (or −Z′′) vs. ω−1/2 plots. Thus, DLi (cm2 s−1) values can be calculated using the following equation:33,53,54
| | |
DLi = R2T2/(2A2n4F4C2σ2)
| (1) |
where
R is the gas constant,
T is the absolute temperature (K),
A is the surface area of the electrode (cm
2),
n is the number of transferred electrons per LiMnPO
4 molecule upon complete delithiation,
F is the Faraday constant,
C is the Li-ion concentration in LiMnPO
4 (0.022 mol cm
−3), and
σ is the Warburg factor (Ω Hz
1/2). The
DLi values of the three samples are listed in
Table 2. We can see that LMP-3.0 has a higher
DLi value than the other samples, implying the quickest Li-ion bulk diffusion. This can explain its better rate capability and enhanced high-rate cycling stability. The EIS results are consistent with the different electrochemical behaviors of the three samples.
4. Conclusions
In summary, we proposed a facile solvothermal route to synthesize LiMnPO4 nanocrystals by using CA as a surfactant. The morphology and size of LiMnPO4 change greatly upon adding a small amount of CA, which realizes the conversion of spindle-like LiMnPO4 of 200 nm into granule-like LiMnPO4 of 30–50 nm. The LiMnPO4 granules display a superior rate capability and cycling stability at high rates after coating with a uniform/thin carbon layer of ∼1 nm thickness. At 20C, a high discharge capacity of 96.6 mA h g−1 can be achieved for LiMnPO4/C. The excellent rate capability is attributed to the small size with easy Li-ion transport at the electrode/electrolyte interface and in the bulk material, and to the uniform/thin carbon layer with enhanced electron transport. The LiMnPO4/C granules also show outstanding high-rate cycling stability, with a discharge capacity of 80.5 mA h g−1 maintained after 500 cycles at 10C. The long cycle life is ascribed to the small size which alleviates the lattice strain, and the uniform carbon coating which prevents Mn dissolution. The superior electrochemical performance of LiMnPO4/C makes it promising for application in EVs.
Acknowledgements
This work was financially supported by the National Basic Research Program of China (2013CB934001), the National Natural Science Foundation of China (No. 51572238), Zhejiang Provincial Natural Science Foundation of China under Grant No. LY15E010004, and the Program for Innovative Research Team in University of Ministry of Education of China (IRT13037).
Notes and references
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188–1194 CrossRef CAS.
- L. X. Yuan, Z. H. Wang, W. X. Zhang, X. L. Hu, J. T. Chen, Y. H. Huang and J. B. Goodenough, Energy Environ. Sci., 2011, 4, 269–284 CAS.
- O. K. Park, Y. Cho, S. Lee, H. C. Yoo, H. K. Song and J. Cho, Energy Environ. Sci., 2011, 4, 1621–1633 Search PubMed.
- C. Delacourt, P. Poizot, M. Morcrette, J. M. Tarascon and C. Masquelier, Chem. Mater., 2004, 16, 93–99 CrossRef CAS.
- M. Yonemura, A. Yamada, Y. Takei, N. Sonoyama and R. Kanno, J. Electrochem. Soc., 2004, 151, A1352–A1356 CrossRef CAS.
- F. Zhou, M. Cococcioni, K. Kang and G. Ceder, Electrochem. Commun., 2004, 6, 1144–1148 CrossRef.
- C. Delacourt, L. Laffont, R. Bouchet, C. Wurm, J. B. Leriche, M. Morcrette, J. M. Tarascon and C. Masqueliera, J. Electrochem. Soc., 2005, 152, A913–A921 CrossRef CAS.
- D. Morgan, A. van der Ven and G. Ceder, Electrochem. Solid-State Lett., 2004, 7, A30–A32 CrossRef CAS.
- A. Yamada, M. Yonemura, Y. Takei, N. Sonoyama and R. Kanno, Electrochem. Solid-State Lett., 2005, 8, A55–A58 CrossRef CAS.
- D. Choi, J. Xiao, Y. J. Choi, J. S. Hardy, M. Vijayakumar, M. S. Bhuvaneswari, J. Liu, W. Xu, W. Wang, Z. G. Yang, G. L. Graff and J. G. Zhang, Energy Environ. Sci., 2011, 4, 4560–4566 CAS.
- N. Meethong, H. Y. Shadow Huang, S. A. Speakman, W. Craig Carter and Y. M. Chiang, Adv. Funct. Mater., 2007, 17, 1115–1123 CrossRef CAS.
- L. F. J. Piper, N. F. Quackenbush, S. Sallis, D. O. Scanlon, G. W. Watson, K. W. Nam, X. Q. Yang, K. E. Smith, F. Omenya, N. A. Chernova and M. S. Whittingham, J. Phys. Chem. C, 2013, 117, 10383–10396 CAS.
- Y. Mishima, T. Hojo, T. Nishio, H. Sadamura, N. Oyama, C. Moriyoshi and Y. Kuroiwa, J. Phys. Chem. C, 2013, 117, 2608–2615 CAS.
- V. Aravindan, J. Gnanaraj, Y. S. Lee and S. Madhavi, J. Mater. Chem. A, 2013, 1, 3518–3539 CAS.
- G. Y. Chen, J. D. Wilcox and T. J. Richardson, Electrochem. Solid-State Lett., 2008, 11, A190–A194 CrossRef CAS.
- P. F. Xiao, B. Ding, M. O. Lai and L. Lu, J. Electrochem. Soc., 2013, 160, A918–A926 CrossRef CAS.
- V. Ramar and P. Balaya, Phys. Chem. Chem. Phys., 2013, 15, 17240–17249 RSC.
- S. Liu, H. S. Fang, E. R. Dai, B. Yang, Y. C. Yao, W. H. Ma and Y. N. Dai, Electrochim. Acta, 2014, 116, 97–102 CrossRef CAS.
- Q. Lu, G. S. Hutchings, Y. Zhou, H. L. Xin, H. M. Zheng and F. Jiao, J. Mater. Chem. A, 2014, 2, 6368–6373 CAS.
- M. S. Kim, J. P. Jegal, K. C. Roh and K. B. Kim, J. Mater. Chem. A, 2014, 2, 10607–10613 CAS.
- K. Kisu, E. Iwama, W. Onishi, S. Nakashima, W. Naoi and K. Naoi, J. Mater. Chem. A, 2014, 2, 20789–20798 CAS.
- L. J. Hu, B. Qiu, Y. G. Xia, Z. H. Qin, L. F. Qin, X. F. Zhou and Z. P. Liu, J. Power Sources, 2014, 248, 246–252 CrossRef CAS.
- X. Zhou, Y. F. Deng, L. N. Wan, X. S. Qin and G. H. Chen, J. Power Sources, 2014, 265, 223–230 CrossRef CAS.
- P. J. Zuo, L. G. Wang, W. Zhang, G. P. Yin, Y. L. Ma, C. Y. Du, X. Q. Cheng and Y. Z. Gao, Nanoscale, 2015, 7, 11509–11514 RSC.
- S. M. Oh, S. W. Oh, C. S. Yoon, B. Scrosati, K. Amine and Y. K. Sun, Adv. Funct. Mater., 2010, 20, 3260–3265 CrossRef CAS.
- F. Wang, J. Yang, P. F. Gao, Y. NuLi and J. L. Wang, J. Power Sources, 2011, 196, 10258–10262 CrossRef CAS.
- Z. H. Qin, X. F. Zhou, Y. G. Xia, C. L. Tang and Z. P. Liu, J. Mater. Chem., 2012, 22, 21144–21153 RSC.
- H. C. Dinh, S. I. Mho, Y. Kang and I. H. Yeo, J. Power Sources, 2013, 244, 189–195 CrossRef CAS.
- S. L. Yang, R. G. Ma, M. J. Hu, L. J. Xi, Z. G. Lu and C. Y. Chung, J. Mater. Chem., 2012, 22, 25402–25408 RSC.
- K. Su, F. Liu and J. T. Chen, J. Power Sources, 2013, 232, 234–239 CrossRef CAS.
- H. Yoo, M. Jo, B. S. Jin, H. S. Kim and J. Cho, Adv. Energy Mater., 2011, 1, 347–351 CrossRef CAS.
- X. H. Rui, X. X. Zhao, Z. Y. Lu, H. T. Tan, D. H. Sim, H. H. Hng, R. Yazami, T. M. Lim and Q. Y. Yan, ACS Nano, 2013, 7, 5637–5646 CrossRef CAS PubMed.
- L. F. Zhang, Q. T. Qu, L. Zhang, J. Li and H. H. Zheng, J. Mater. Chem. A, 2014, 2, 711–719 CAS.
- L. G. Wang, P. J. Zuo, G. P. Yin, Y. L. Ma, X. Q. Cheng, C. Y. Du and Y. Z. Gao, J. Mater. Chem. A, 2015, 3, 1569–1579 CAS.
- W. X. Zhang, Z. Q. Shan, K. L. Zhu, S. Z. Liu, X. Y. Liu and J. H. Tian, Electrochim. Acta, 2015, 153, 385–392 CrossRef CAS.
- Y. Hong, Z. L. Tang and Z. T. Zhang, Electrochim. Acta, 2015, 176, 369–377 CrossRef CAS.
- Y. Hong, Z. L. Tang, S. T. Wang, W. Quan and Z. T. Zhang, J. Mater. Chem. A, 2015, 3, 10267–10274 CAS.
- J. N. Zhu, W. C. Li, F. Cheng and A. H. Lu, J. Mater. Chem. A, 2015, 3, 13920–13925 CAS.
- H. Guo, C. Y. Wu, L. H. Liao, J. Xie, S. C. Zhang, P. Y. Zhu, G. S. Cao and X. B. Zhao, Inorg. Chem., 2015, 54, 667–674 CrossRef CAS PubMed.
- Q. B. Xia, T. Liu, J. J. Xu, X. Y. Cheng, W. Lu and X. D. Wu, J. Mater. Chem. A, 2015, 3, 6301–6305 CAS.
- J. G. Zheng, C. C. Qin, T. F. Wu, S. F. Xie, L. Ni, M. Y. Peng, Y. F. Tang and Y. F. Chen, J. Mater. Chem. A, 2015, 3, 15299–15306 CAS.
- J. G. Zheng, L. Ni, Y. W. Lu, C. C. Qin, P. X. Liu, T. F. Wu, Y. F. Tang and Y. F. Chen, J. Power Sources, 2015, 282, 444–451 CrossRef CAS.
- S. F. Yang, P. Y. Zavalij and M. S. Whittingham, Electrochem. Commun., 2001, 3, 505–508 CrossRef CAS.
- M. K. Devaraju and I. Honma, Adv. Energy Mater., 2012, 2, 284–297 CrossRef CAS.
- D. Choi, D. H. Wang, I. T. Bae, J. Xiao, Z. M. Nie, W. Wang, V. V. Viswanathan, Y. J. Lee, J. G. Zhang, G. L. Graff, Z. G. Yang and J. Liu, Nano Lett., 2010, 10, 2799–2805 CrossRef CAS PubMed.
- G. Y. Chen, A. K. Shukla, X. Y. Song and T. J. Richardson, J. Mater. Chem., 2011, 21, 10126–10133 RSC.
- Y. S. Yu, C. Kim, D. A. Shapiro, M. Farmand, D. N. Qian, T. Tyliszczak, A. L. David Kilcoyne, R. Celestre, S. Marchesini, J. Joseph, P. Denes, T. Warwick, F. C. Strobridge, C. P. Grey, H. Padmore, Y. S. Meng, R. Kostecki and J. Cabana, Nano Lett., 2015, 15, 4282–4288 CrossRef CAS PubMed.
- S. S. Jan, S. Nurgul, X. Q. Shi, H. Xia and H. Pang, Electrochim. Acta, 2014, 149, 86–93 CrossRef CAS.
- X. Tang, S. S. Jan, Y. Y. Qian, H. Xia, J. F. Ni, S. V. Savilov and S. M. Aldoshin, Sci. Rep., 2015, 5, 11958 CrossRef PubMed.
- J. P. Schmidt, T. Chrobak, M. Ender, J. Illig, D. Klotz and E. Ivers Tiffée, J. Power Sources, 2011, 196, 5342–5348 CrossRef CAS.
- J. Illig, M. Ender, T. Chrobak, J. P. Schmidt, D. Klotz and E. Ivers Tiffée, J. Electrochem. Soc., 2012, 159, A952–A960 CrossRef CAS.
- J. Illig, J. P. Schmidt, M. Weiss, A. Weber and E. Ivers Tiffée, J. Power Sources, 2013, 239, 670–679 CrossRef CAS.
- K. S. Lee, K. J. Lee, Y. S. Kang, T. J. Shin, Y. E. Sung and D. Ahn, Nanoscale, 2015, 7, 13860–13867 RSC.
- H. J. Zhu, W. Zhai, M. Yang, X. M. Liu, Y. C. Chen, H. Yang and X. D. Shen, RSC Adv., 2014, 4, 25625–25632 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra21264b |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.