
Polylactide–hydroxyapatite nanocomposites with highly improved interfacial adhesion via mussel-inspired polydopamine surface modification
Hongyang Suna,
Miao Aia,
Siqi Zhua,
Xiaolong Jiaa,
Qing Cai*ab and
Xiaoping Yangab
aState Key Laboratory of Organic-Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, P. R. China
bBeijing Laboratory of Biomedical Materials, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: caiqing@mail.buct.edu.cn; Fax: +86 10 64412884
First published on 19th October 2015
Abstract
The poor interfacial adhesion between organic and inorganic components remains the primary obstacle in obtaining composite scaffolds of high performance for bone tissue engineering. Mussel-inspired dopamine surface modification on inorganic components is a potential solution for this problem. Herein, hydroxyapatite (HA) nano-rods were freshly made by a co-precipitation method and subjected to polydopamine (PDA) coating. Then the modified HA nano-rods were mixed into biodegradable poly(L-lactide) (PLLA) to get PLLA/HA nanocomposites. The PDA modification was found to be mild and easy to handle, and was effective in improving the dispersibility of HA nano-rods in chloroform and especially in PLLA/chloroform solution. The resulting PLLA/HA composite films and porous scaffolds demonstrated significant enhancements in their mechanical properties at relatively high contents (30–60 wt%) of modified HA nano-rods in comparison with those composites containing unmodified HA nano-rods. This was thought to be mainly attributed to both the even distribution of modified HA nano-rods throughout the PLLA matrix and the strong interfacial adhesion between HA and PLLA components. The PLLA/HA composites displayed good biocompatibility with bone mesenchymal stem cells (BMSCs) and could enhance the osteogenic differentiation of BMSCs, indicating the PDA modification has no adverse effect on biological properties. These results confirmed the idea of using mussel-inspired dopamine surface modification as a feasible and efficient approach in developing organic–inorganic composite materials for bone regeneration studies.
1. Introduction
It is well known that natural bone tissues have a kind of complex and hierarchical structure consisting of components such as inorganic nano-hydroxyapatite (nano-HA) and organic collagen.1 Benefiting from their good biocompatibility, bioactivity and osteoconductivity, stoichiometric nano-HAs are widely used as bioceramic fillers in polymeric matrices for developing bone restoration substitutes.2–6 In the case of bone tissue engineering, the applications of collagen or biodegradable aliphatic polyesters containing nano-HAs are well known and intensively investigated.7 Aggregation of nano-HAs, however, is usually observed in composites due to the high surface energy of nano-HAs and the inherently poor phase compatibility between inorganic nanoparticles and organic matrices. This often leads to low mechanical strength of the resulting composites.8,9 In particular, tensile strengths of composite films decrease rapidly as the content of nano-HAs increases.10–12 And thus, the substrates are not strong enough for supporting bone regeneration by using the strategy of tissue engineering.To obtain good dispersion of nano-HAs in the collagen matrix, Teng et al.13 modified the preparation of the collagen/nano-HA solution by mixing freshly-made nano-HA precipitates with collagen solution. In the cases of aliphatic polyesters being applied, these were mainly polylactone-based homopolymers and copolymers, however, various surface modification methods have been attempted on nano-HAs to improve their interfacial adhesion with polymeric biomaterials. Silane coupling agents were firstly used as a surface modifier for nano-HAs because of their simple and quick treatment steps.14,15 The chemical bonds between the nano-HAs and the polymeric matrix were enhanced to some extent. However, the organic matrices with different chemical structures indicated silane coupling agent selection, and the dispersion of nano-HAs in polyesters was still not satisfactory. Based on the principles of similarity and intermiscibility, the surface grafting of polyester oligomers onto nano-HAs has been widely studied in the last decade. Hong et al.12 and Li et al.16 modified nano-HAs by grafting polylactide (PLA) via the ring-opening polymerization of lactide using the hydroxyls on the HA surface as initiators. Lee et al.17 and Hu et al.18 coupled poly(ε-caprolactone) (PCL) or poly(lactide-co-glycolide) (PLGA) onto nano-HAs by the reaction between the hydroxyls of HA and the end-groups of polyesters. In our previous work,19 acrylate-ended PLA oligomers were grafted onto nano-HAs via atom transfer radical polymerization (ATRP). These approaches were found effective in ameliorating the interfacial compatibility between nano-HAs and the corresponding polyester matrices. However, these methods could be seen as somewhat complicated and toxic reagents were likely involved. Besides, the surface grafted polymeric chains could only achieve an acceptable improvement in interfacial adhesion in polymeric matrices with similar molecular structures. The mechanical properties of the resulting nanocomposites usually began to decline when the added amounts of nano-HAs were above 5–10 wt%.12,16 To obtain applicable substrates for bone tissue engineering, therefore, it was still very necessary to look for more effective and efficient, as well as simpler modification methods for nano-HAs.
In recent years, scientists found that 3,4-dihydroxy-L-phenylalanine (DOPA) played an important role for mussels in forming an extraordinarily robust adhesion on boats and rocks etc.20–22 Dopamine (DA), an analogue of DOPA, is thus widely applied to modify various substrates including metals,23,24 oxides,25,26 carbon materials27–29 and polymers,30–32 benefiting from its adhesive properties and its oxidative polymerization features in aqueous solution. The coating process is simple and mild, causing no adverse effect on the structure of the original substrate. In the area of biomedical applications, polydopamine (PDA) surface modification was usually applied to enhance cell adhesion, proliferation and differentiation.33–35 These publications confirmed the PDA component was biocompatible and non-cytotoxic, making it a suitable surface modifier for biomaterials.
With respect to using PDA modification to improve interfacial adhesion in composites, some reports have emerged in the last few years. Yang et al.36 constructed a surface layer of PDA on clay, and dispersed the modified clay into epoxy resin. It was suggested that the hydrogen bonding between PDA and epoxy resin macromolecules had enhanced the effective interfacial stress transfer between different phases, leading to improvement in the mechanical properties of the resulting composites. Song et al.37 reported that barium titanate (BaTiO3) nanoparticles and nanofibers were perfect dielectric fillers for epoxy composites after surface modification with PDA. The explanation was the stronger interfaces between the filler and the resin giving rise to higher breakdown strength. Similar modifications were conducted on boehmite nanoparticles38 and carbon fibers,39 and both the modified fillers demonstrated good affinity to epoxy resin.
With these approaches, it was highly expected that PDA coating on nano-HAs might enhance the interfacial adhesion between nano-HAs and biodegradable polyesters, and thus increase the dispersibility of nano-HAs in polyesters and improve the mechanical properties of the resulting composites. To this end, HA nano-rods were prepared via a chemical precipitation method and treated with DA aqueous solution in this study. The dispersibility of the modified nano-HAs (m-HA) in solvent was evaluated by standing observation and particle size distribution. Composites containing poly(L-lactide) (PLLA) and different amounts of m-HAs were fabricated into thin films or porous scaffolds, and characterizations including morphological observations and mechanical properties measurements were conducted. To determine the effect of PDA modification on PLLA/m-HA composites, untreated nano-HAs were used as the reference. Finally, in vitro cell culture with bone mesenchymal stem cells (BMSCs) was carried out to evaluate the potential of PLLA/m-HA composites for use in bone regeneration.
2. Experimental
2.1. Materials
PLLA (Mw = 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was purchased from Purac (Netherlands). Dopamine hydrochloride and Tris were purchased from Sigma and used directly. Reagents for the preparation of nano-HAs, i.e. calcium nitrate tetrahydrate (Ca(NO3)2·4H2O), diammonium hydrogen phosphate ((NH4)2HPO4) and ammonium hydroxide (NH3·H2O, 25%) were bought from Aldrich. Solvents and other chemicals involved in this study were of analytically pure grade and obtained from Beijing Chemical Plant (China).
000) was purchased from Purac (Netherlands). Dopamine hydrochloride and Tris were purchased from Sigma and used directly. Reagents for the preparation of nano-HAs, i.e. calcium nitrate tetrahydrate (Ca(NO3)2·4H2O), diammonium hydrogen phosphate ((NH4)2HPO4) and ammonium hydroxide (NH3·H2O, 25%) were bought from Aldrich. Solvents and other chemicals involved in this study were of analytically pure grade and obtained from Beijing Chemical Plant (China).
2.2. Synthesis of nano-HAs
The nano-HAs used in this study were freshly synthesized using a wet chemical precipitation method for the following surface modification.19,40 Briefly, Ca(NO3)2·4H2O as a calcium source and (NH4)2HPO4 as a phosphorus source were dissolved individually in distilled water at the Ca/P molar ratio of 1.67. The initial pH of each solution was adjusted to 10 with NH3·H2O. Then the (NH4)2HPO4 solution was dropped into the Ca(NO3)2·4H2O solution, which was thermoset at 90 °C, followed by continuous stirring for 3 h. At the end of the reaction, the suspension was centrifuged and the precipitates were washed three times with distilled water. Finally, the obtained nano-HAs were kept wet with a spot of water for further use. For characterizations, the products were freeze-dried thoroughly.2.3. Modification on nano-HAs with PDA coating
The wet nano-HAs (0.2 g) were re-dispersed in 100 mL Tris solution (10 mM) with pH being adjusted to 8.5 with HCl solution. The suspensions were ultrasonically (500 W) treated for 5 min to ensure the good dispersion of nano-HAs. Dopamine hydrochloride was then added into the system to obtain a DA aqueous solution of 2.0 mg mL−1. The coating reaction was performed at room temperature with continuous magnetic stirring for 48 h. Subsequently, the modified nano-HAs were washed with distilled water three times and freeze-dried.2.4. Preparation of PLLA/HA films
To prepare PLLA/HA composite films, the solution-casting method was applied. Briefly, modified or unmodified nano-HA powders were dispersed in chloroform under magnetic stirring for 24 h with concentrations of nano-HAs from 0.01 to 0.15 g mL−1 and ultrasonically treated (500 W, 5 min) before mixing. Then 5 mL of either of the above HA-containing suspensions was added into 5 mL of chloroform solution containing 0.1 g mL−1 of PLLA, to achieve HA contents from 10 to 60 wt% in reference to PLLA weight. The mixture solutions were cast onto glass plates, standing for 48 h at room temperature to allow for solvent evaporation. Then the films were further vacuum-dried at 37 °C to constant weight to remove any residual solvent. The pure PLLA film was prepared in a similar way without the addition of nano-HAs.2.5. Preparation of PLLA/HA porous scaffolds
To prepare PLLA/HA porous scaffolds, a combination of thermal-induced phase separation (TIPS) and particle-leaching methods were applied.41 Briefly, anhydrous and sieved NaCl particles (150–300 μm) were mixed into the aforementioned mixture solutions containing PLLA and nano-HAs (10, 30 or 60 wt%). To perform the TIPS, dioxane was used instead of chloroform in this operation. Slurry was obtained as the weight ratio of NaCl to PLLA was 20:1. The slurry was then cast into a cylinder mold (ϕ = 10 mm, h = 50 mm), followed by freezing in liquid nitrogen and freeze-drying for 24 h. After that, the composites were taken out from the mold and soaked in distilled water for 24 h to leach out the salt with the water being refreshed every 6 h. The porous composites were freeze-dried for another 24 h and sponge-like foams were obtained. For comparison, pure PLLA porous scaffolds were also prepared in a similar way.
2.6. Characterizations
Chemical compositions of the prepared samples were determined by X-ray photoelectron spectroscopy (XPS), Fourier transform infrared spectroscopy (FT-IR) and Raman spectroscopy measurements. XPS was performed on an XPS spectrometer (ESCALAB 250, USA) with a monochromatized Al Kα X-ray source (1486.6 eV photons) under vacuum (8–10 Torr) using an incidence angle of 45° at a power of 150 W. FT-IR was recorded on a spectrometer (Nicolet 8700, USA) with wavenumbers ranging from 4000 to 400 cm−1 at a resolution of 4 cm−1, where samples were milled with potassium bromide powder and pressed into transparent tablets. Raman measurement was performed at 633 nm by using a Raman spectroscope (Renishaw inVia, UK). The amount of PDA coating on nano-HAs was determined by thermogravimetric analysis (TGA) using a Q50 thermogravimetric analyzer (TA instruments, USA) in an air atmosphere from room temperature to 700 °C at a heating rate of 20 °C min. The crystalline structures of the produced nano-HAs and m-HAs were evaluated by wide-angle X-ray diffraction (WAXD) from 20° to 80° using an Ultima III X-ray diffractometer with a Cu tube anode (2500VB2+/PC, 40 kV, 200 mA). The produced nano-HAs and m-HAs were observed under a transmission electron microscope (TEM, Hitachi 800) to determine their dispersibility and morphology by dispersing samples in chloroform for 24 h and dropping onto 800-mesh copper grids. For size distribution analysis, dynamic light scattering (DLS) was carried out by using a BI-90Plus (Brookhaven, USA) with a solid laser (35 mW). Morphological observations of nano-HAs, PLLA/HA composite films and scaffolds were conducted by a scanning electron microscope (SEM, S-4700, Hitachi) at an accelerating voltage of 20 kV after being sputter-coated with platinum (30 mA, 20 s) using a sputter-coater (Polaron E5600, USA). Calcium element mapping was performed under the same parameters as SEM observation with an exposure time of 180 s. The tensile properties of different samples were determined with an Instron 1211 machine by applying a 50 N load cell at a stretching speed of 10 mm min−1 according to the standard ISO 527-3:1995. Each sample was cut into dumbbell-shaped specimens with effective dimensions of 20 mm in length and 4 mm in width. The thickness of each specimen was measured by a digital thickness meter. The storage compressive modulus (E′) of porous scaffold slices (ϕ = 10 mm, h = 2.5 mm) was monitored using dynamic mechanical thermal analysis (DMTA, TA Q800), with loading frequency changing from 0.1 Hz to the occurrence of resonance, under a fixed deformation of 5%.2.7. Biological evaluations
Rat BMSCs (purchased from Cell Culture Center, Peking Union Medical College, China) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Hyclone) supplemented with 10% fetal bovine serum (FBS, PAA, Germany), 100 IU per mL penicillin (Sigma), and 100 mg mL−1 streptomycin (Sigma). The culture was maintained with 5% CO2 at 37 °C and saturated humidity until 80% confluence prior to use. For osteogenic differentiation assays, 0.05 mmol L−1 vitamin C (Sigma), 10 mmol L−1 b-sodium glycerophosphate (Sigma), and 1 × 10−8 mol L−1 dexamethasone (Sigma) were added to the culture medium.2.8. Statistical analysis
The results of the tensile test and the compressive test were represented as mean ± standard deviation for n = 5 and all biological quantitative data were represented as mean ± standard deviation for n = 3. Statistical analysis was made based on t-tests and a difference between groups of *p ≤ 0.05 was considered significant and **p ≤ 0.01 was considered highly significant.3. Results and discussion
3.1. nano-HA synthesis and modification
HA nanoparticles used for PDA coating modification were freshly prepared in the lab by using a wet chemical precipitation method. In comparison with commercial nano-HA powders, which normally display significant aggregation in re-dispersing, the as-prepared wet nano-HAs were considered able to disperse better in the DA aqueous solution to guarantee the surface coating efficiency. The obtained precipitates from the mixture solution of (NH4)2HPO4 and Ca(NO3)2·4H2O were firstly characterized by using XPS, FT-IR, XRD and SEM to confirm the formation of nano-HAs. As shown in Fig. 1a, the XPS spectrum of nano-HAs revealed the presence of calcium, phosphorus and oxygen elements, which were essential components of calcium phosphate compounds. A weak signal assigned to carbon was found, which was thought to originate from the absorbed carbon dioxide on the surface of nano-HAs. | ||
| Fig. 1 Characterizations of nano-HAs prepared from chemical precipitation method before (nano-HA) and after PDA coating (m-HA): (a) XPS spectra; (b) enlarged N1s XPS signal from (a); (c) FT-IR spectra; (d) Raman spectra; (e) TGA curves; and (f) XRD patterns. | ||
The corresponding FT-IR spectrum of nano-HAs (Fig. 1c) displayed a standard profile of HA with characteristic absorption peaks at 565 cm−1, 604 cm−1, 1026 cm−1 and 1090 cm−1, representing the presence of PO43− groups.42 In XRD characterization (Fig. 1f), a typical pattern of HA crystalline structure was identified, as the characteristic diffraction of (0 0 2), (2 1 0), (2 1 1), (3 0 0), (2 0 2), (3 1 0), (2 2 2), (2 1 3) and (4 1 1) crystal faces was clearly detected.19 The morphological observations of as-prepared nano-HAs are shown in Fig. 2a, c and e. Rod-like particles, with ∼180 nm in length and ∼15 nm in width, were clearly observed. Under TEM observation (Fig. 2e), the surface of the as-prepared nano-HAs could be seen to be quite smooth with no hint of any surface coating. The aggregation detected in Fig. 2c was thought due to the sample preparation for TEM observation, in which the nano-HA precipitates were freeze-dried and re-dispersed in chloroform.
 | ||
| Fig. 2 Morphological observations of nano-HAs prepared from the chemical precipitation method before (left column) and after PDA coating (right column) using SEM (a, b) and TEM (c–f). Images (e) and (f) are magnifications of images (c) and (d), respectively. | ||
To perform the PDA coating modification, the freshly prepared wet nano-HAs were directly dispersed in DA aqueous solution without the drying step. At the end of the coating reaction, the initially white nano-HAs turned dark-brown, and were retrieved from the solution, freeze-dried and systematically characterized to identify the formation of the PDA surface layer. In comparison with the original nano-HAs, a new peak assigned to nitrogen was detected, in addition, the signal intensity of the carbon element was clearly enhanced in the XPS spectrum of m-HAs in addition to the signals of calcium, phosphorus and oxygen elements (Fig. 1a and b). The XPS spectrum of m-HAs demonstrated a combination of the signals of pure PDA and nano-HAs. Similarly, as shown in Fig. 1c, the FT-IR spectrum of m-HAs also possessed the characteristic absorption peaks of both PDA and HA. The peaks at 1602 cm−1 and at 1515 cm−1 were ascribed to the overlap of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C resonance vibration in aromatic rings and the N–H bending and shearing vibration in PDA.43 In Fig. 1f, the XRD pattern of m-HAs resembled that of nano-HAs well, indicating no change in crystalline structure taking place after the coating modification. To further identify the success of PDA coating on nano-HAs, Raman spectrum and TGA measurements were also applied. As shown in Fig. 1d, the spectrum of m-HA resembled the profile of PDA, although weaker in signal strength, showing two peaks at 1409 cm−1 and 1576 cm−1 assigned to the aromatic group in PDA.38,44,45 With all these facts, the successful PDA coating on nano-HAs was confirmed. From TGA data (Fig. 1e), the decomposition of PDA was seen taking place when the heating temperature was above 300 °C, with almost no residual at 700 °C. Meanwhile, nano-HAs demonstrated high thermal stability in the temperature range of 300–500 °C, and a final weight loss of 8 wt% was determined due to the elimination of moisture and other possible residuals. In the case of m-HAs, obvious weight loss was detected as the temperature increased, achieving a leveled-off weight loss of 24 wt% at the temperature of 700 °C. In comparison, the amount of the coated PDA on nano-HAs could be identified as about 16 wt%. With the surface PDA coating, the morphology of m-HAs slightly changed from the original nano-HAs. In comparison with the original nano-HAs, as the SEM image shows (Fig. 2b), polymer-like materials could be seen clearly on or between HA nano-rods (Fig. 2a). From the enlarged TEM image (Fig. 2f), a layer of several nanometers in thickness was identified.
C resonance vibration in aromatic rings and the N–H bending and shearing vibration in PDA.43 In Fig. 1f, the XRD pattern of m-HAs resembled that of nano-HAs well, indicating no change in crystalline structure taking place after the coating modification. To further identify the success of PDA coating on nano-HAs, Raman spectrum and TGA measurements were also applied. As shown in Fig. 1d, the spectrum of m-HA resembled the profile of PDA, although weaker in signal strength, showing two peaks at 1409 cm−1 and 1576 cm−1 assigned to the aromatic group in PDA.38,44,45 With all these facts, the successful PDA coating on nano-HAs was confirmed. From TGA data (Fig. 1e), the decomposition of PDA was seen taking place when the heating temperature was above 300 °C, with almost no residual at 700 °C. Meanwhile, nano-HAs demonstrated high thermal stability in the temperature range of 300–500 °C, and a final weight loss of 8 wt% was determined due to the elimination of moisture and other possible residuals. In the case of m-HAs, obvious weight loss was detected as the temperature increased, achieving a leveled-off weight loss of 24 wt% at the temperature of 700 °C. In comparison, the amount of the coated PDA on nano-HAs could be identified as about 16 wt%. With the surface PDA coating, the morphology of m-HAs slightly changed from the original nano-HAs. In comparison with the original nano-HAs, as the SEM image shows (Fig. 2b), polymer-like materials could be seen clearly on or between HA nano-rods (Fig. 2a). From the enlarged TEM image (Fig. 2f), a layer of several nanometers in thickness was identified.
The surface coating of PDA increased the dispersibility of HA nano-rods. Qualitatively, the scattered m-HA nano-rods shown in Fig. 2d demonstrated much better dispersibility than the aggregations of original nano-HAs shown in Fig. 2c. In standing stability evaluation, the original nano-HAs and m-HAs were dispersed in chloroform and left standing for 0.5 h and 4 h, respectively. As shown in Fig. 3e and f, the original nano-HAs completely descended to the bottom of the bottle within 0.5 h, while m-HAs could stay suspended for a longer time. However, in pure chloroform, m-HAs still descended slowly to the bottom of the bottle within several hours. In view of the goal of this study being to prepare PLLA/HA nanocomposites, m-HAs were further dispersed in PLLA solution in chloroform and left standing. Interestingly, m-HAs demonstrated excellent dispersion and stability in the PLLA solution without any obvious hint of descending after standing for 4 h.
 | ||
| Fig. 3 Analysis of particle size and particle size distribution of original nano-HAs (a) and PDA-coated nano-HAs (m-HAs) (b–d) by DLS, together with macroscopical observation of HA sedimentation in both chloroform and PLLA/chloroform solutions after standing for 0.5 h (e) and 4 h (f). The DLS data in (b–d) indicates the particle size and particle size distribution of m-HAs after they were dispersed in chloroform for 0.5 h (b), 24 h (c) and 72 h (d). | ||
Quantitatively, particle sizes and particle size distributions of original nano-HAs and m-HAs in chloroform were measured by DLS. As shown in Fig. 3a, the size distribution of the original nano-HAs was quite wide and the particle size was very large. Two groups of particles with average sizes of ∼900 nm and ∼3500 nm were detected. This indicated that the unmodified nano-HAs were liable to aggregate in chloroform, which was consistent with the observation that the original nano-HAs descended quickly in chloroform (Fig. 3e). The data in Fig. 3b were measured immediately after m-HAs were dispersed in chloroform, i.e. within 0.5 h after the suspension was prepared. It could be seen the particle size of m-HAs was smaller and their particle size distribution was narrower than those of the original nano-HAs. This suggested that the PDA coating on the HA surface had hindered particle aggregation and improved the dispersibility of m-HAs in chloroform. The primary explanation for this improvement was thought to be due to the swelling feature of PDA macromolecules in the solvent. To verify this inference, the suspension containing m-HAs and chloroform was magnetically stirred for a further 24 h and 72 h, and DLS measurement was conducted again. As shown in Fig. 3c and d, the amount of particles at an average particle size of 300–400 nm was reduced with longer mixing time. At the same time, the particle size distribution could be seen becoming much narrower with longer mixing time. At 72 h after mixing, the average particle size was almost uniform around 687 nm with quite narrow distribution. This change in particle size and particle size distribution of m-HAs in chloroform along with time was attributed to the gradual swelling of the PDA coating layer on the HA surface, and the PDA chains achieved a fully stretched state by being soaked in chloroform for 3 days. The final particle size was the HA nanoparticle with its swollen PDA surface layer, accordingly, the aggregation of nano-HAs was prevented and a uniform particle size was detected. Therefore, the standing stability of m-HAs in chloroform was improved in comparison with the original nano-HAs, as shown in Fig. 3e and f. However, sedimentation of m-HAs in chloroform could still be observed because the density of HA (3.16 g cm−3) was much higher than that of chloroform (1.48 g cm−3). If m-HAs were dispersed in PLLA/chloroform solution, the extended PDA chains on the HA surface could interact with PLLA chains, which are able to prevent m-HAs descending in the PLLA/chloroform solution. As shown in Fig. 3f, the suspension of m-HAs in PLLA/chloroform was still quite stable after standing for 4 h. This finding was welcome in the following preparations of PLLA/HA composite films and porous scaffolds.
3.2. PLLA/HA composite films
To prepare PLLA/HA composite films, original or PDA modified nano-HAs were ultrasonically dispersed in PLLA/chloroform solution and cast onto glass plates to allow for solvent evaporation. After the films were vacuum-dried to constant weight, SEM observations and tensile properties evaluations were conducted to detect the effects of PDA modification and nano-HA amount on film performance. The morphology of PLLA/HA composite films containing different amounts of original nano-HAs or m-HAs is presented in Fig. 4. In the case of original nano-HAs being incorporated, aggregations of nano-HAs became more and more significant as their amounts increased from 10 wt% to 60 wt%, as shown in Fig. 4a–c. On the contrary, the m-HAs could be seen dispersing quite well in the PLLA matrix with no obvious hint of aggregation when the contents of m-HAs were 10 wt% (Fig. 4d) and 20 wt% (Fig. 4e). As the incorporated content of m-HAs was further increased to 30–60 wt%, m-HAs were still dispersed quite evenly throughout the PLLA matrix (Fig. 4f–i). When the content of m-HAs was above 40 wt%, the composites displayed a morphology of HA nanoparticles being bonded homogeneously together by the PLLA matrix, and thus rough surfaces were observed. The significant difference in the dispersibility between the original nano-HAs and PDA modified m-HAs apparently came from the interaction between the swelling PDA macromolecules on the m-HA surface and the PLLA chains in the solution state, which led to PLLA/m-HA composite films with even m-HA distribution. | ||
| Fig. 4 Morphological observations of PLLA/HA composite films containing different contents of the original nano-HAs or m-HAs: (a) nano-HAs, 10 wt%; (b) nano-HAs, 20 wt%; (c) nano-HAs, 60 wt%; (d) m-HAs, 10 wt%; (e) m-HAs, 20 wt%; (f) m-HAs, 60 wt%; (g) m-HAs, 30 wt%; (h) m-HAs, 40 wt%; (i) m-HAs, 50 wt%. | ||
The tensile properties of these PLLA/HA composite films are shown and compared in Fig. 5. In comparison with the pure PLLA film, the addition of only 10 wt% of the original nano-HAs caused a slight decrease in the tensile strength of the PLLA/HA composite film (Fig. 5a). Accordingly, the elongation at break of the PLLA/HA composite film was also decreased in comparison with the pure PLLA film (Fig. 5b). This is a common disadvantage of using unmodified nano-HAs as fillers for polymeric matrices,10–12,15 because significant aggregations of nano-HAs are unavoidable due to their poor interfacial compatibility with polymeric components. In the case of m-HAs, however, it could be envisioned that both the decrease in HA aggregation and the improvement in interfacial adhesion between m-HA and PLLA would be able to ameliorate the mechanical properties of the resulting PLLA/m-HA composites. As shown in Fig. 5a, the tensile strength of PLLA/m-HA composite films gradually increased as the incorporation content of m-HAs increased from 0 wt% to 30 wt%, obtaining an increase of 50% at a content of 30 wt% in comparison with the pure PLLA film. The elongations at break of these composite films were also found to be slightly higher than that of the pure PLLA film (Fig. 5b). Apparently, the PDA on the nano-HA surface not only improved the interfacial adhesion between the HA nano-rods and PLLA matrix to give higher tensile strength, but also had a plasticizing effect on PLLA to result in longer elongation ratios. When the content of m-HAs was higher than 30 wt%, however, decreases in both tensile strength and elongation at break were detected for the PLLA/m-HA composite films, and they decreased continuously as the content of m-HAs increased from 30 wt% to 60 wt%, even with the distribution of m-HAs in the PLLA matrix being uniform. It was not surprising to have this finding, because the content of PLLA (i.e. the binding component) was relatively reduced as the content of m-HAs increased. The composite films in this study were prepared by solution-casting from mixture solutions, which were completely different from the case of natural bone tissues, which possess high mechanical properties at high contents of HA due to the assembled collagen–HA structure.1,9
 | ||
| Fig. 5 Tensile strength (a) and tensile stress–strain curves (b) of PLLA/HA composite films containing different contents of original nano-HAs or m-HAs. | ||
3.3. PLLA/HA composite scaffolds
For bone tissue engineering, the mechanical properties of PLLA/HA porous composite scaffolds are quite essential for bone regeneration, especially for load-bearing bone repair applications. Herein, PLLA porous scaffolds were fabricated by introducing original nano-HAs or PDA-coated m-HAs via similar particle-leaching and TIPS techniques. As shown in Fig. 6, the pure PLLA and PLLA/HA composite scaffolds displayed similar porous structure, with average pore size of 150–300 μm resulting from particle-leaching. The homogeneous porous structures on pore walls were resulting from TIPS. | ||
| Fig. 6 SEM images of cross-sections of porous PLLA and PLLA/HA scaffolds containing 10 wt% of original nano-HAs or PDA-coated m-HAs: (a, b) pure PLLA; (c, d) PLLA/nano-HA; (e, f) PLLA/m-HA. The arrows in image (d) point to the aggregations of nano-HAs. | ||
At lower magnification, the composite scaffolds containing 10 wt% of original nano-HAs or m-HAs did not show much difference in morphology (Fig. 6a, c and e). Due to the rough cross-sections, no obvious aggregations of original nano-HAs could be seen on the pore walls (Fig. 6c). At larger magnification, however, significant aggregations of nano-HAs were clearly detected, which were embedded in the porous structures on pore walls as indicated by arrows (Fig. 6d). Quite differently, no such aggregations or even a hint of HA particles could be detected in the case of 10 wt% of m-HAs being introduced into the PLLA scaffold (Fig. 6f). Higher contents of m-HAs were thus introduced into PLLA scaffolds. As shown in Fig. 7, the addition of 30 wt% and 60 wt% of m-HAs did not alter the dual porous architecture of PLLA/m-HA composite scaffolds. Some HA particles were detected on pore walls until the content of m-HAs reached 60 wt%. To confirm the existence of HA components in the PLLA/m-HA composite scaffolds, calcium element mapping was conducted on pore walls. As shown in Fig. 7, calcium was detected clearly in all three of the PLLA/m-HA composite scaffolds, and the signal strength apparently increased as the content of m-HA increased. Considering the results of Fig. 6 and 7, it was undoubted that the PDA-coated m-HAs had achieved an excellent and uniform dispersion in the PLLA porous scaffold, even at a high content of 60 wt%, which was much better than that of the original nano-HAs.
 | ||
| Fig. 7 SEM images and corresponding calcium element mapping of cross-sections of porous PLLA/HA scaffolds containing 10 wt% (a, b, b′), 30 wt% (c, d, d′) and 60 wt% (e, f, f′) of m-HAs. | ||
All the aforementioned porous scaffolds were submitted to compressive properties evaluation by using DMTA. Storage compressive modulus (E′) against loading frequency was monitored from 0.1 Hz to the occurrence of resonance and is presented in Fig. 8. The E′ of pure PLLA scaffold was the lowest among all the scaffolds. With the addition of 10 wt% of original nano-HAs, the E′ value of the corresponding PLLA/HA composite scaffold increased slightly, although the dispersion of nano-HAs was poor (Fig. 6d). The improvement in compressive properties was suggested to be due to the rigid feature of the HA component. As for the PLLA/m-HA composite scaffolds, their compressive properties were greatly enhanced by introducing PDA-coated m-HAs, demonstrating gradual increase in E′ values along with higher contents of m-HAs. Besides, the PLLA/m-HA composite scaffold demonstrated a higher E′ value than the PLLA/nano-HA composite scaffold when they had the same content of HA component (10 wt%). Therefore, the inorganic rigid HA particles were able to reinforce the PLLA scaffold, enhancing its compressive properties, and the enhancement was more efficient if the two phases had good interfacial adhesion. Effective interfacial stress transfer was readily achieved upon compression with the m-HAs having strong interfacial interactions with the PLLA matrix via their PDA surface layer.
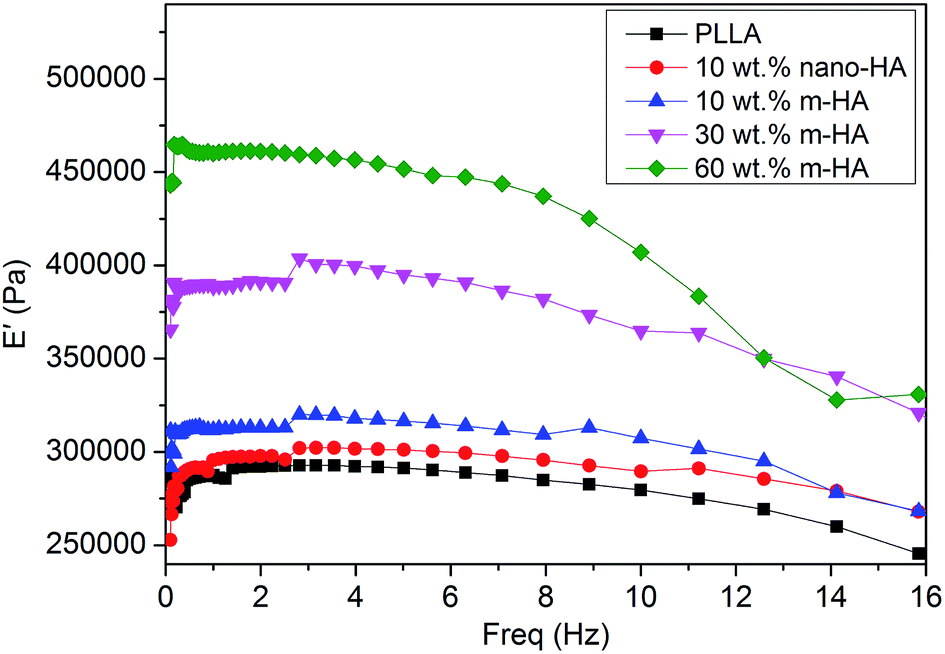 | ||
| Fig. 8 DMTA evaluation of the storage compressive modulus (E′) against loading frequency for porous PLLA and PLLA/HA scaffolds containing different amounts of original nano-HAs or m-HAs. | ||
3.4. Biological properties
Targeting bone regeneration, the biological properties of the resulting PLLA/m-HA scaffolds were evaluated in terms of cytotoxicity (Fig. 9), cell viability (Fig. 10) and osteogenic differentiation (Fig. 11). According to ISO standard, extracts from different scaffolds (pure PLLA, PLLA/HA containing modified or unmodified nano-HAs) were made and used for BMSCs culture. As shown in Fig. 9, BMSCs proliferated continuously in all the extracts along with culture time, which was comparable with the negative control and much faster than the positive control. From Fig. 10, the BMSCs seeded onto all the scaffolds could be seen growing well with time, as the increasing numbers of fluorescent stained cells show. Live cells were stained green, and red staining spots for dead cells could hardly be found at all time points on all scaffolds. These results definitely indicated the non-cytotoxicity of PLLA and PLLA/HA composite scaffolds to BMSCs. Instead, they could support the proliferation of BMSCs.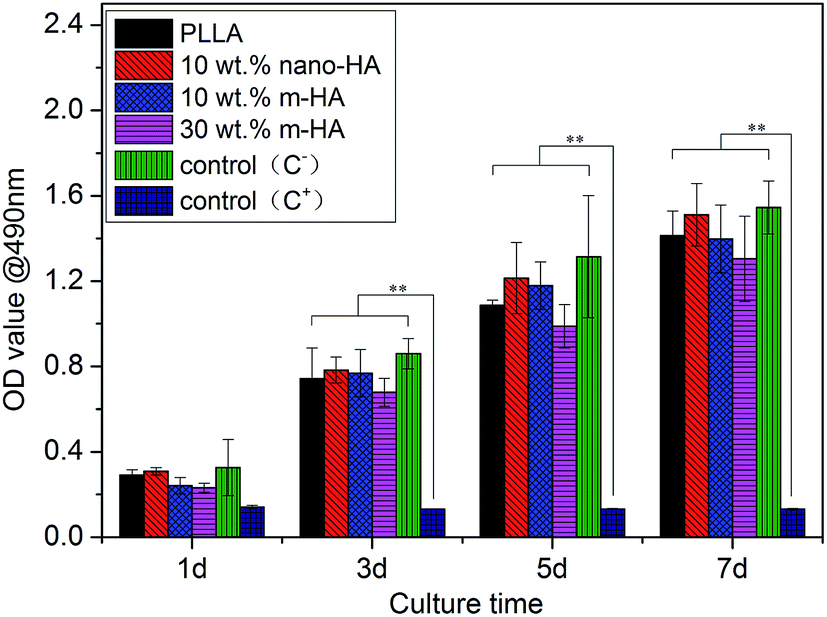 | ||
| Fig. 9 Proliferation of BMSCs in extracts of PLLA and PLLA/HA composite scaffolds containing unmodified (nano-HA) or PDA-modified nano-HAs (m-HA), using DMEM and phenol as negative and positive control, respectively (**P < 0.01). | ||
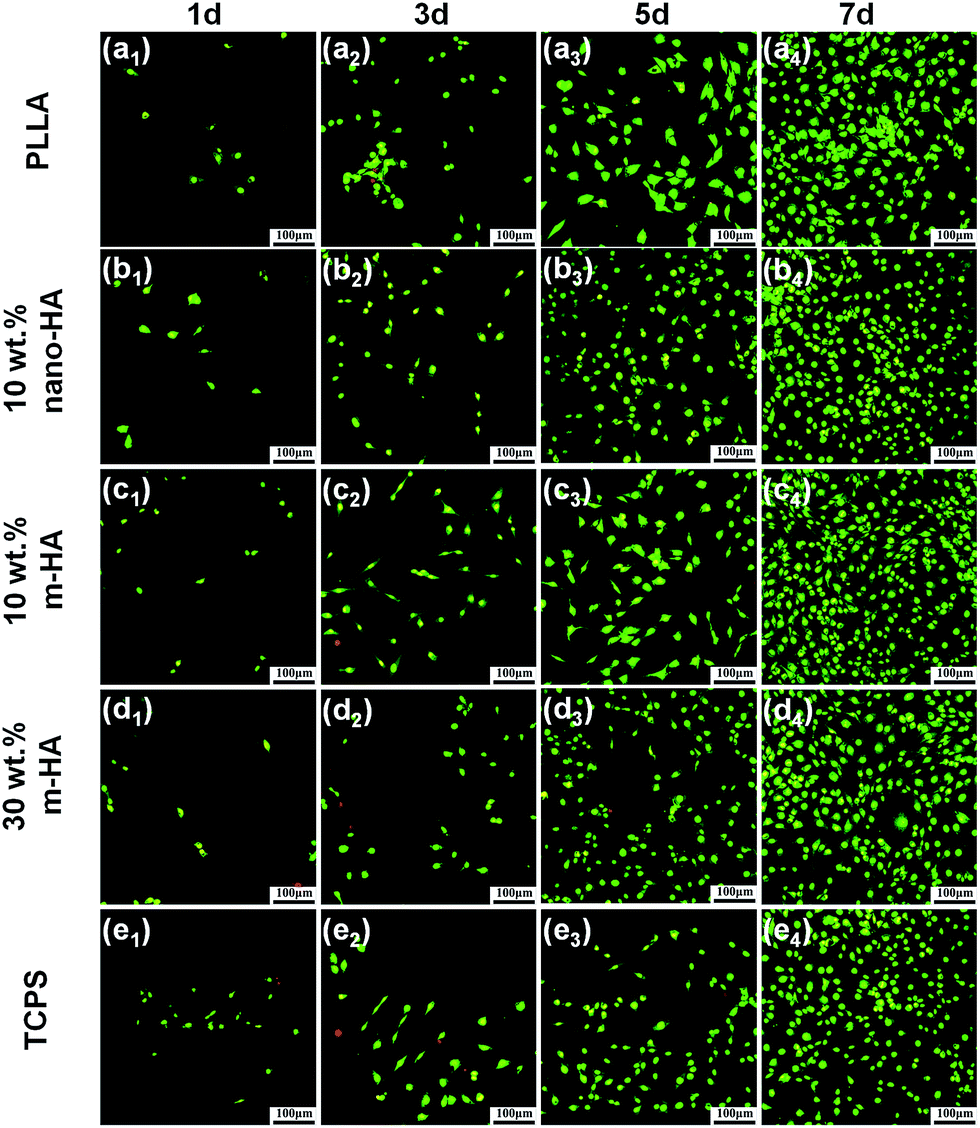 | ||
| Fig. 10 Live/dead assay of BMSCs proliferated on PLLA and PLLA/HA composite scaffolds containing unmodified (nano-HA) or PDA-modified nano-HAs (m-HA) for 1, 3, 5 and 7 days by AO/EB staining (green: live; red: dead), using TCPS as control. | ||
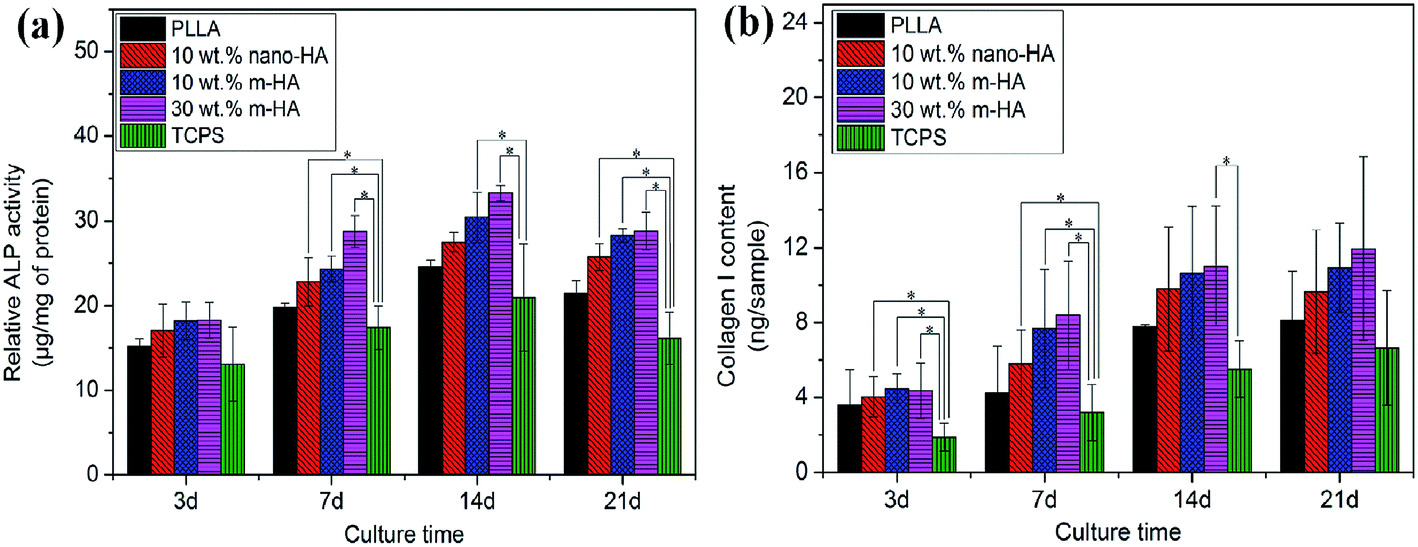 | ||
| Fig. 11 ALP activity (a) and Col-I content (b) of BMSCs cultured on PLLA and PLLA/HA composite scaffolds containing unmodified (nano-HA) or PDA-modified nano-HAs (m-HA) at different time points, using TCPS as control (*p < 0.05). | ||
One interesting thing found in Fig. 9 was that BMSCs seemingly proliferated slightly slower in the extracts from PLLA/m-HA composite scaffolds, especially in the sample containing 30 wt% of m-HA, although they did not have a significant difference from other scaffolds. It is suggested that the dissolution of HA components into the extracts caused the difference in cell proliferation rates, because the calcium and phosphate ions were bioactive to enhance osteogenic differentiation.46,47 In comparison with unmodified nano-HA aggregations, as shown in the scaffold morphology in Fig. 6 and 7, the well-dispersed m-HAs in composite scaffolds might facilitate the dissolution of HA components faster and more easily due to their better exposure to the medium. Accordingly, it was proposed the osteogenic differentiation of BMSCs on different scaffolds might be in a reverse order to the cell proliferation.
As shown in Fig. 11, both the ALP and Col-I expression were much higher on all porous scaffolds than on TCPS, indicating the significant promotion of osteogenic differentiation of BMSCs on scaffolds. It was obviously benefiting from the 3D porous structure of the scaffolds in comparison with the 2D flat TCPS surface.48,49 With the incorporation of nano-HAs, acceleration in the osteogenic differentiation of BMSCs was further identified in comparison with the pure PLLA scaffold due to the bioactivity of the HA component. Noticeably, PLLA/m-HA scaffolds displayed stronger ability in enhancing the osteogenic differentiation of BMSCs than PLLA/nano-HA scaffolds, even when they contained the same amount of HA component. Compared to the latter, obviously, it was the modified HA nano-rods dispersed homogeneously throughout the PLLA/m-HA scaffold that created a favorable environment for the growth and differentiation of BMSCs. As more m-HAs were incorporated, the enhancement of osteogenic differentiation was further promoted.
All in all, with their non-cytotoxicity and ability to enhance osteogenic differentiation, the PLLA/m-HA composite scaffolds were envisioned to be good substrates for bone regeneration.
4. Conclusion
Surface modification of nano-HAs using DA aqueous solution soaking was efficient and effective in improving the interfacial adhesion between nano-HA and the PLLA matrix. The PDA coating procedure was mild and simple, and did not cause adverse effects on the HA structure. The PDA surface layer demonstrated a swelling feature in chloroform, and thus modified nano-HAs could disperse well and remain stable in PLLA/chloroform solution via the interaction of PDA macromolecules and PLLA chains. PLLA/HA composite films and porous scaffolds containing a high content of m-HAs were thus readily fabricated to achieve good mechanical properties. The PDA modification did not cause adverse effects on cell viability, proliferation and differentiation. This study thus suggests a universal surface modification approach for nano-HAs, which could have wide applications in bone tissue engineering by replacing PLLA with other biodegradable biomaterials.Acknowledgements
The authors acknowledge the financial support from National Basic Research Program of China (2012CB933904), National Natural Science Foundation of China (No. 51473016 and 51373016), and Beijing Municipal Commission of Education (ZDZH20141001001).Notes and references
- J.-Y. Rho, L. Kuhn-Spearing and P. Zioupos, Med. Eng. Phys., 1998, 20, 92 CrossRef CAS PubMed.
- H. J. Zhou and J. Lee, Acta Biomater., 2011, 7, 2769 CrossRef CAS PubMed.
- J. R. Woodard, A. J. Hilldore, S. K. Lan, C. J. Park, A. W. Morgan, J. A. Eurell, S. G. Clark, M. B. Wheeler, R. D. Jamison and A. J. Wagoner Johnson, Biomaterials, 2007, 28, 45 CrossRef CAS PubMed.
- H. Deplaine, J. L. G. Ribelles and G. G. Ferrer, Compos. Sci. Technol., 2010, 70, 1805 CrossRef CAS.
- F. Mohandes and M. Salavati-Niasari, RSC Adv., 2014, 4, 25993 RSC.
- J. J. Guan, J. Yang, J. Q. Dai, Y. H. Qin, Y. Wang, Y. P. Guo, Q. F. Ke and C. Q. Zhang, RSC Adv., 2015, 5, 36175 RSC.
- J. Venkatesan and S.-K. Kim, J. Biomed. Nanotechnol., 2014, 10, 3124 CrossRef CAS PubMed.
- M. Wang, Biomaterials, 2003, 24, 2133 CrossRef CAS PubMed.
- M. R. Rogel, H. J. Qiu and G. A. Ameer, J. Mater. Chem., 2008, 18, 4233 RSC.
- X. F. Song, F. G. Ling, L. L. Ma, C. G. Yang and X. S. Chen, Compos. Sci. Technol., 2013, 79, 8 CrossRef CAS.
- H. X. Diao, Y. F. Si, A. P. Zhu, L. J. Ji and H. C. Shi, Mater. Sci. Eng., C, 2012, 32, 1796 CrossRef CAS.
- Z. K. Hong, X. Y. Qiu, J. R. Sun, M. X. Deng, X. S. Chen and X. B. Jing, Polymer, 2004, 45, 6699 CrossRef CAS.
- S. H. Teng, E. J. Lee, P. Wang and H. E. Kim, Mater. Lett., 2008, 62, 3055–3058 CrossRef CAS.
- X. J. Wang, G. J. Song and T. Lou, Med. Eng. Phys., 2010, 32, 391 CrossRef PubMed.
- Z. Fang and Q. L. Feng, Mater. Sci. Eng., C, 2014, 35, 190 CrossRef CAS PubMed.
- J. Li, X. L. Lu and Y. F. Zheng, Appl. Surf. Sci., 2008, 255, 494 CrossRef CAS.
- H. J. Lee, H. W. Choi, K. J. Kim and S. C. Lee, Chem. Mater., 2006, 18, 5111 CrossRef CAS.
- X. X. Hu, H. Shen, F. Yang, X. J. Liang, S. D. Wang and D. C. Wu, Appl. Surf. Sci., 2014, 292, 764 CrossRef CAS.
- J. Q. He, X. P. Yang, J. F. Mao, F. J. Xu and Q. Cai, Appl. Surf. Sci., 2012, 258, 6823 CrossRef CAS.
- F. Bernsmann, V. Ball, F. Addiego, A. Ponche, M. Michel, J. J. Gracio, V. Toniazzo and D. Ruch, Langmuir, 2011, 27, 2819 CrossRef CAS PubMed.
- Q. Lin, D. Gourdon, C. Sun, N. Holten-Andersen, T. H. Anderson, J. H. Waite and J. N. Israelachvili, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3782 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426 CrossRef CAS PubMed.
- X. H. Yu, J. Walsh and M. Wei, RSC Adv., 2013, 4, 7185 RSC.
- L. Zhang, S. Yuan, S. Chen, D. R. Wang, B. Z. Han and Z. M. Dang, Compos. Sci. Technol., 2015, 110, 126 CrossRef CAS.
- E. Mazario, J. Sánchez-Marcos, N. Menéndez, P. Herrasti, M. García-Hernández and A. Muñoz-Bonilla, RSC Adv., 2014, 4, 48353 RSC.
- M. Zhang, J. Zheng, Y. Zheng, J. L. Xu, X. W. He, L. X. Chen and Q. L. Fang, RSC Adv., 2013, 3, 13818 RSC.
- H. Y. Hu, B. Yu, Q. Ye, Y. S. Gu and F. Zhou, Carbon, 2010, 48, 2347 CrossRef CAS.
- T. H. Yoon and Y. J. Park, RSC Adv., 2014, 4, 17434 RSC.
- J. Luo, N. Zhang, R. Liu and X. Y. Liu, RSC Adv., 2014, 4, 64816 RSC.
- V. K. Thakur, D. Vennerberg, S. A. Madbouly and M. R. Kessler, RSC Adv., 2014, 4, 6677 RSC.
- B. P. Tripathi, N. C. Dubey, F. Simon and M. Stamm, RSC Adv., 2014, 4, 34073 RSC.
- R. Sa, Z. H. Wei, Y. Yan, L. Wang, W. C. Wang, L. Q. Zhang, N. Y. Ning and M. Tian, Compos. Sci. Technol., 2015, 113, 54 CrossRef CAS.
- S. R. Nielsen, F. Besenbacher and M. Chen, Phys. Chem. Chem. Phys., 2013, 15, 17029 RSC.
- S. H. Ku, J. Ryu, S. K. Hong, H. Lee and C. B. Park, Biomaterials, 2010, 31, 2535 CrossRef CAS PubMed.
- Y. Li, Y. Z. Shi, S. Duan, D. Y. Shan, Z. P. Wu, Q. Cai and X. P. Yang, J. Biomed. Mater. Res., Part A, 2014, 102, 3894 CrossRef PubMed.
- L. P. Yang, S. L. Phua, J. K. Teo, C. L. Toh, S. K. Lau, J. Ma and X. H. Lu, ACS Appl. Mater. Interfaces, 2011, 3, 3026 CAS.
- Y. Song, Y. Shen, H. Y. Liu, Y. H. Lin, M. Li and C.-W. Nan, J. Mater. Chem., 2012, 22, 16491 RSC.
- Y.-C. Chung and J.-Y. Huang, Thin Solid Films, 2014, 570, 376 CrossRef CAS.
- W. Lee, J. U. Lee and J.-H. Byun, Compos. Sci. Technol., 2015, 110, 53 CrossRef CAS.
- P. P. Wang, C. H. Li, H. Y. Gong, X. R. Jiang, H. Q. Wang and K. X. Li, Powder Technol., 2010, 203, 315 CrossRef CAS.
- L. Q. Zheng, F. Yang, H. Shen, X. F. Hu, C. Mochizuki, M. Sato, S. G. Wang and Y. D. Zhang, Biomaterials, 2011, 32, 7053 CrossRef CAS PubMed.
- L. Y. Jiang, C. D. Xiong, D. L. Chen, L. X. Jiang and X. B. Pang, Appl. Surf. Sci., 2012, 259, 72 CrossRef.
- Z. Y. Xi, Y. Y. Xu, L. P. Zhu, Y. Wang and B. K. Zhu, J. Membr. Sci., 2009, 327, 244 CrossRef CAS.
- Y. H. Sun, Y. Deng, Z. Y. Ye, S. S. Liang, Z. H. Tang and S. C. Wei, Colloids Surf., B, 2013, 111, 107 CrossRef CAS PubMed.
- N. G. Rim, S. J. Kim, Y. M. Shin, I. Jun, D. W. Lim, J. H. Park and H. Shin, Colloids Surf., B, 2012, 91, 189 CrossRef CAS PubMed.
- Q. Lei, J. Chen, W. X. Huang, D. Wu, H. Z. Lin and Y. Z. Lai, Chem.-Biol. Interact., 2015, 233, 139–146 CrossRef CAS PubMed.
- A. Singh and J. Elisseeff, J. Mater. Sci., 2010, 20, 8832 CAS.
- X. F. Qiu, Y. T. Zhang, X. Z. Zhao, S. W. Zhang, J. H. Wu, H. Q. Guo and Y. Q. Hu, Biomaterials, 2015, 53, 600–608 CrossRef CAS PubMed.
- H. Shen, Y. F. Ma, Y. Luo, X. Y. Liu, Z. J. Zhang and J. W. Dai, Colloids Surf., B, 2015, 135, 332–338 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |