
Catalytic dehydrochlorination of 1,2-dichloroethane to produce vinyl chloride over N-doped coconut activated carbon†
Abstract
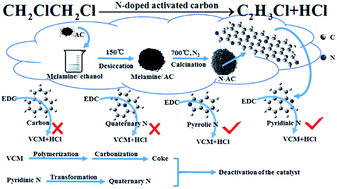
A series of N-doped coconut activated carbon catalysts (N-AC) were prepared using melamine as the nitrogen precursor and their performance for the catalytic dehydrochlorination of 1,2-dichloroethane (1,2-DCE) to produce vinyl chloride monomer (VCM) was assessed. It is indicated that the N-doped catalyst 5 : 10-N-AC exhibits a stable catalytic activity at 250 °C with the 1,2-DCE conversion of 85.1% at 180 h. Through DFT calculations, it is suggested that both pyridinic and pyrrolic nitrogen dopants can adsorb preferentially 1,2-DCE and increase the activity for 1,2-DCE dehydrochlorination at low temperature, in combination with characterizations of BET, Raman, TG, TPD, XPS, etc. In addition, the increase of quaternary nitrogen dopants and coking deposition on the catalyst surface can result in the activity decline. The N-doped activated carbon catalyst provides a promising pathway to produce VCM through a low-temperature energy-saving process of 1,2-DCE catalytic dehydrochlorination.
 Please wait while we load your content...
Please wait while we load your content...

