
Fatty acyl incorporation in the biosynthesis of WAP-8294A, a group of potent anti-MRSA cyclic lipodepsipeptides†
Abstract
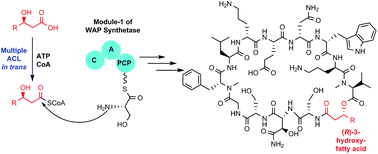
WAP-8294A is a family of at least 20 cyclic lipodepsipeptides exhibiting potent anti-MRSA activity. These compounds differ mainly in the hydroxylated fatty acyl chain; WAP-8294A2, the most potent member of the family that reached clinical trials, is based on (R)-3-hydroxy-7-methyloctanoic acid. It is unclear how the acyl group is incorporated because no acyl-CoA ligase (ACL) gene is present in the WAP-8294A gene cluster in Lysobacter enzymogenes OH11. Here, we identified seven putative ACL genes in the OH11 genome and showed that the yield of WAP-8294A2 was impacted by multiple ACL genes with the ACL6 gene having the most significant effect. We then investigated several (R)-3-hydroxy fatty acids and their acyl SNAC (N-acetylcysteamine) thioesters as substrates for the ACLs. Feeding (R)-3-hydroxy-7-methyloctanoate-SNAC to the ACL6 gene deletion mutant restored the production of WAP-8294A2. Finally, we heterologously expressed the seven ACL genes in E. coli and purified six of the proteins. While these enzymes exhibit a varied level of activity in vitro, ACL6 showed the highest catalytic efficiency in converting (R)-3-hydroxy-7-methyloctanoic acid to its CoA thioester when incubated with coenzyme A and ATP. These results provided both in vivo and in vitro evidence to support the fact that ACL6 is the main player for fatty acyl activation and incorporation in WAP-8294A2 biosynthesis. The results also suggest that the molecular basis for the acyl chain diversity in the WAP-8294A family is the presence of functionally overlapping ACLs.
 Please wait while we load your content...
Please wait while we load your content...

