
Influence of Na+ ion doping on the phase change and upconversion emissions of the GdF3: Yb3+, Tm3+ nanocrystals obtained from the designed molecular precursors†
Abstract
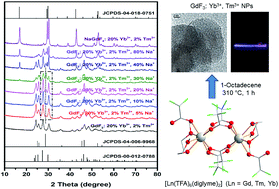
Bottom-up synthesis of a series of GdF3 nanocrystals (NCs) co-doped with 20 mol% Yb3+, 2 mol% Tm3+ and varying amount of Na+ ions is reported using novel molecular precursors [Ln2(TFA)6(diglyme)2] [Ln = Gd (1), Tm (2), Yb (3)] and [Na4(TFA)4(diglyme)]∞ (4) [TFA = CF3COO−; diglyme = MeO(CH2CH2O)2Me]. The single crystal X-ray structures of the new complexes 1–4, which act as excellent precursors because of the absence of water molecules and the ability of the diglyme ligand to behave as a capping reagent during decomposition to render the nanoparticles monodisperse in organic solvents, show a versatile bonding mode of the TFA (dangling η1 or bridging μ,η1,η1; μ4-η1:η1:η1:η1- and μ4-η1:η1:η1:η2 (O, F)) and diglyme (η3 or μ-η3:η1) ligands. The influence of Na+ ion doping on the phase change and upconversion emissions of these GdF3: Yb3+, Tm3+ NCs was studied and compared with the upconversion (UC) intensity of the well-known upconverting NaGdF4: Yb3+, Tm3+ NCs. Among the several UC samples studied, the GdF3: Yb3+, Tm3+ NCs with 30 mol% Na+ seemed to be the most promising as a red emitting UC phosphor, emitting more photons than the NaGdF4: Yb3+, Tm3+ UC NCs in the near IR region.
 Please wait while we load your content...
Please wait while we load your content...

