DOI:
10.1039/C5RA20760F
(Paper)
RSC Adv., 2015,
5, 97874-97882
Arene-based fluorescent probes for the selective detection of iron†
Received
7th October 2015
, Accepted 4th November 2015
First published on 9th November 2015
Abstract
Pyridine-2,6-dicarboxamide based probes containing phenyl (L1), naphthyl (L2) and anthracenyl (L3) groups have been synthesized and screened towards Na+, Mg2+, Ca2+, Cr3+, Mn2+, Mn3+, Fe2+, Fe3+, Co2+, Ni2+, Cu2+, Zn2+, Hg2+, Cd2+ and Pb2+ ions. Probes L2 and L3 exhibited remarkable sensing for Fe2+ and Fe3+ ions with L3 being particularly selective. Collective studies comprising of Job's plot, Benesi–Hildebrand fitting, Stern–Volmer plot and detection limit display notable sensing ability of probe L3 for Fe3+ as compared to Fe2+ ion. Competitive binding studies in presence of other metal ions further illustrated the high efficiency of L3. The probe L3 binds to Fe3+ ion by forming a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex with the apparent association constant (Ka) of 3.31 × 103 M−1. L3–Fe3+ system is shown to have potential applications in logic gate and cell imaging.
:1 complex with the apparent association constant (Ka) of 3.31 × 103 M−1. L3–Fe3+ system is shown to have potential applications in logic gate and cell imaging.
Introduction
Development of sensors for transition metal ions has received a great deal of attention due to relevance in a wide range of environmental and biological processes.1,2 Iron is one of the physiologically most abundant transition metals in the living organism including humans. An average human adult contains approximately 3–5 g of iron and the total cellular concentration is approximately 50–100 μM.3 Iron is involved in numerous cellular processes such as electron-transport, oxygen uptake, metabolism, and transcriptional regulation.4,5 Biological iron almost exclusively exists in the ferrous (Fe2+) and ferric (Fe3+) state, although other oxidation states are routinely stabilized during catalytic cycles.1a Cells must carefully control iron levels, distribution, and speciation as excessive Fe3+ ion is responsible for several diseases including cancer, hepatitis, hemochromatosis, and dysfunction of vital organs.6,7 Therefore, detection of iron especially in ferrous versus ferric form is immensely important and fluorescent probes are attractive tools to visualize its concentration and distribution.8
Many probes for iron exhibit “turn off” fluorescence response towards its binding.9 However, a probe must be able to distinguish between the two oxidation states of iron. Because of the propensity of Fe2+ to oxidize to Fe3+ form under aerobic aqueous environment, it has been a challenge to design probes specific for ferrous ion.1a In last few years, several probes have been developed for the selective response to either Fe2+ or Fe3+ form or simply iron species.9–18 Although most of these probes have only been used for modest applications in living cells, further developments including highly selective probes will undoubtedly result in better insights into iron cellular biology.
Scaffolds based on pyridine-2,6-dicarboxamide fragment have been widely used for anion sensing abilities.19,20 Such a noteworthy feature is due to the desirable orientation of the hydrogen bonds in such scaffolds which assist in anion recognition. On the other hand, such scaffolds have also been used for coordinating assorted metal ions with notable structural and functional applications.21,22 In this work, we have used three pyridine-2,6-dicarboxamide based probes (L1, L2 and L3) displaying the presence of a chelating cavity while also incorporating appended phenyl, naphthyl and anthracenyl groups as the fluorophores. Such fluorescent probes have been used for the selective binding of iron while displaying no interference from other metal ions. We also illustrate the application of one such probe in logic gate and in cell imaging.
Results and discussion
Synthesis and characterization of probes
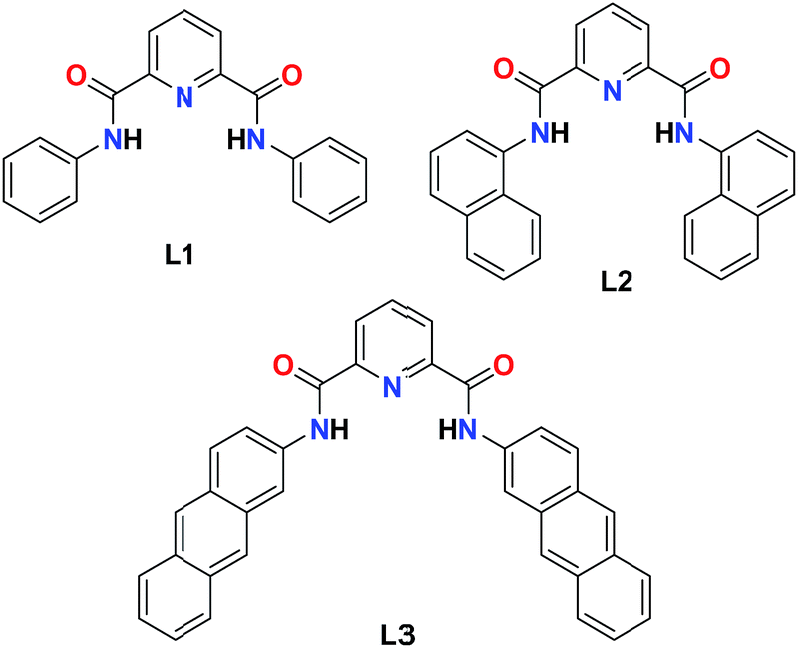
We have synthesized three pyridine-2,6-dicarboxamide based probes containing phenyl (L1), naphthyl (L2) and anthracenyl (L3) groups (Scheme 1). Probes L1 and L2 were synthesized according to the reported procedures23,24 whereas L3 was synthesized by the coupling of pyridine-2,6-dicarboxylic acid with 2-aminoanthracene. FTIR spectrum of L3 shows N–H peak at 3270 cm−1 while Oamide stretch was noted at 1657 cm−1 (Fig. S1, ESI†). The 1H NMR spectrum shows N–H resonance at 11.3 ppm while aromatic protons appeared between 7.4 and 8.9 ppm (Fig. S2, ESI†). The ESI+ mass spectrum of probe L3 displays the molecular ion peak at 518.1868 for [L3 + H+] (Fig. S3, ESI†). The probes L1, L2, and L3 display absorption spectral maxima in THF at 300, 312 and 310 nm, respectively (Fig. S4, ESI†).
 |
| | Scheme 1 Schematic representations of probes L1, L2 and L3. | |
Fluorescence studies
The photophysical properties of probes L1, L2 and L3 were examined in various solvents such as THF, MeOH, MeCN, DMF and CH2Cl2. Probe L1 showed a very poor emission at ca. 420 nm upon excitation at 300 nm at a very high concentration of 100 μM in MeOH (Fig. S5, ESI†). Probe L2 exhibited a prominent emission at 408 nm after excitation at 335 nm (Fig. S6, ESI†) whereas probe L3 displayed a significant emission at 440 nm after excitation at 340 nm (Fig. S7, ESI†). Notably, both L2 and L3 displayed similar results in several solvents. It is clear that the presence of naphthyl and anthacenyl moieties in L2 and L3 is the major source of emission whereas phenyl ring in L1 is not an effective flurophore.
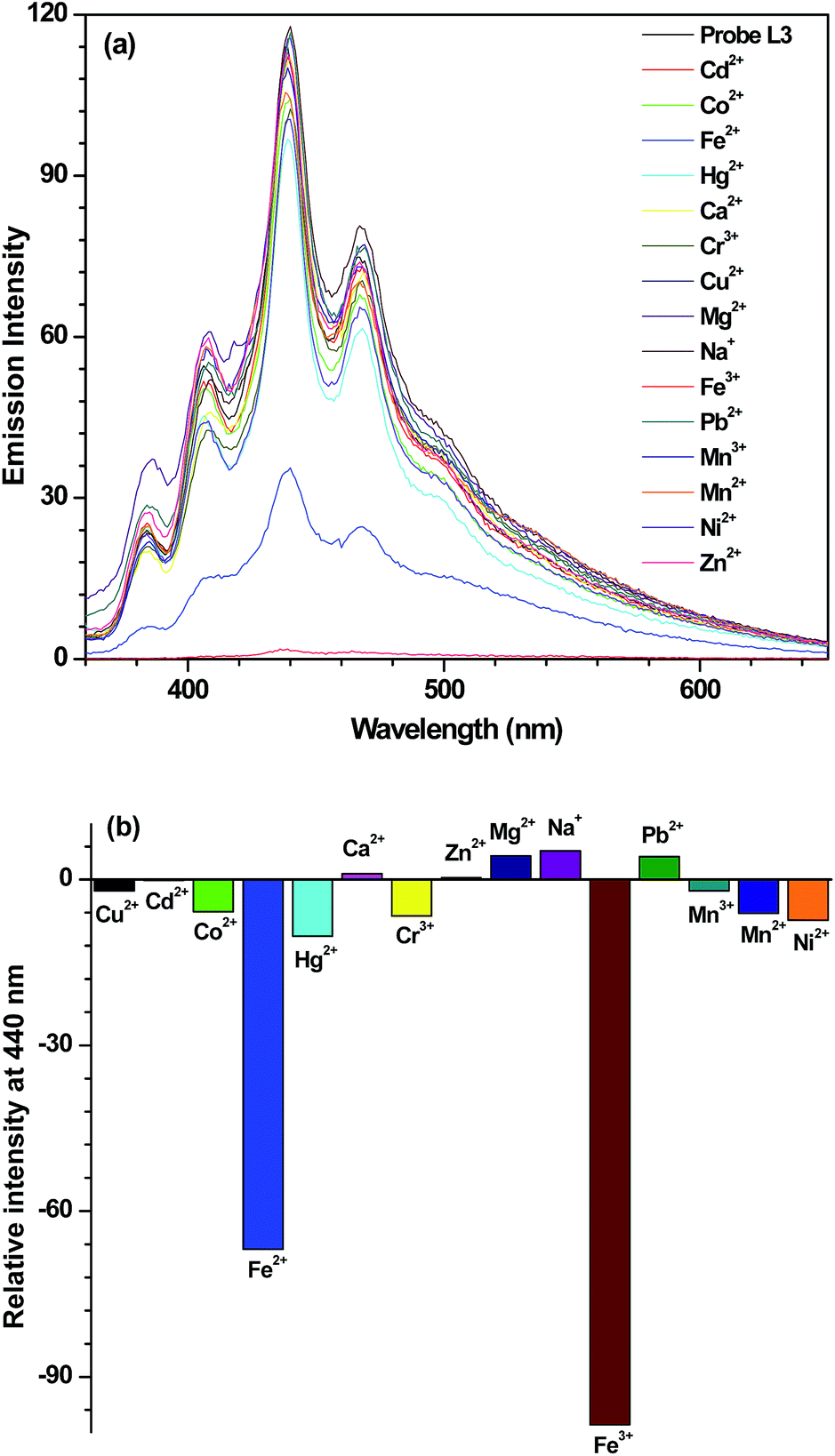
Addition of 10 equivalents of assorted metal ions, such as Na+, Mg2+, Ca2+, Cr3+, Mn2+, Mn3+, Co2+, Ni2+, Cu2+, Zn2+, Hg2+, Cd2+ and Pb2+, did not cause any appreciable change in the emission intensity of probe L2 (Fig. S8a, ESI†) and L3 (Fig. 1a). On the other hand, introduction of Fe2+ and Fe3+ ion resulted in considerable quenching in the emission intensity (Fig. S8b (ESI)† and 1b). A comparison of two probes, L2 and L3, illustrate that although both are selective for Fe2+ and Fe3+ ions; selectivity towards Fe3+ state is quite high (Fig. 2). Further, out of two probes, the performance of L3 is superior than that of L2.
 |
| | Fig. 1 (a) Emission spectrum of probe L3 (40 μM) in THF and after its interaction with 10 equiv. of assorted metal ions. (b) Bar diagram exhibiting change in emission intensity of probe L3 (40 μM) in THF after the addition of 10 equiv. of assorted metal ions. | |
 |
| | Fig. 2 Bar diagram exhibiting change in emission intensity of L2 (40 μM) and L3 (40 μM) in THF after addition of 10 equiv. of metal ions. | |
We then investigated the fluorescence quenching for both probes L2 and L3 by the incremental addition of 1–10 equivalents of Fe2+ and Fe3+ ions. For probe L2, addition of 10 equivalents of Fe2+ causes 62% quenching while Fe3+ resulted in 91% quenching (Fig. 3a). Importantly, probe L3 displayed nearly quantitative quenching on the addition of 10 equivalents of Fe3+whereas a maximum of 65% quenching was noted for Fe2+ion (Fig. 3b). To determine the extent of fluorescence quenching of probes L2 and L3 by Fe2+ and Fe3+ ions; Stern–Volmer constants (KSV) were calculated.25 The Stern–Volmer plots for probes L2 and L3 are shown in Fig. 3c and d, respectively whereas KSV are presented in Table 1. The higher values of KSV indicate strong quenching of probe L2 and L3 by Fe3+ ion when compared to Fe2+ ion. A further comparison exhibits a strong sensing ability of probe L3 towards Fe3+ ion as compared to Fe2+ ion. Detection limits were also calculated both for L2 and L3 and the values are presented in Table 1 (see Fig. S9 and S10, ESI†).26 Both probes L2 and L3 showed good and excellent detection limits for Fe2+ and Fe3+ ions, respectively. It is clear from the Stern–Volmer constants and detection limits that the probe L3 shows a very high selectivity towards Fe3+ ion as compared to L2. Therefore, probe L3 was selected for further studies.
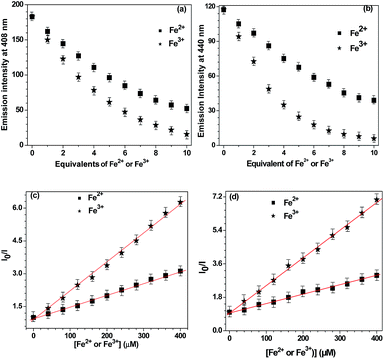 |
| | Fig. 3 Change in emission intensity with number of equivalents of Fe2+ versus Fe3+ ions for probe L2 (a) and L3 (b). Stern–Volmer plots for Fe2+ versus Fe3+ ions for probe L2 (c) and L3 (d). The points represent the mean ± the standard error of three independent sets. | |
Table 1 Stern–Volmer constants (KSV) and detection limits
| Species |
KSV (M−1) |
Detection limit (μM) |
| L2 + Fe2+ |
5.49 × 103 |
08.54 |
| L2 + Fe3+ |
1.33 × 104 |
06.08 |
| L3 + Fe2+ |
4.94 × 103 |
11.27 |
| L3 + Fe3+ |
1.48 × 104 |
05.56 |
Selectivity studies
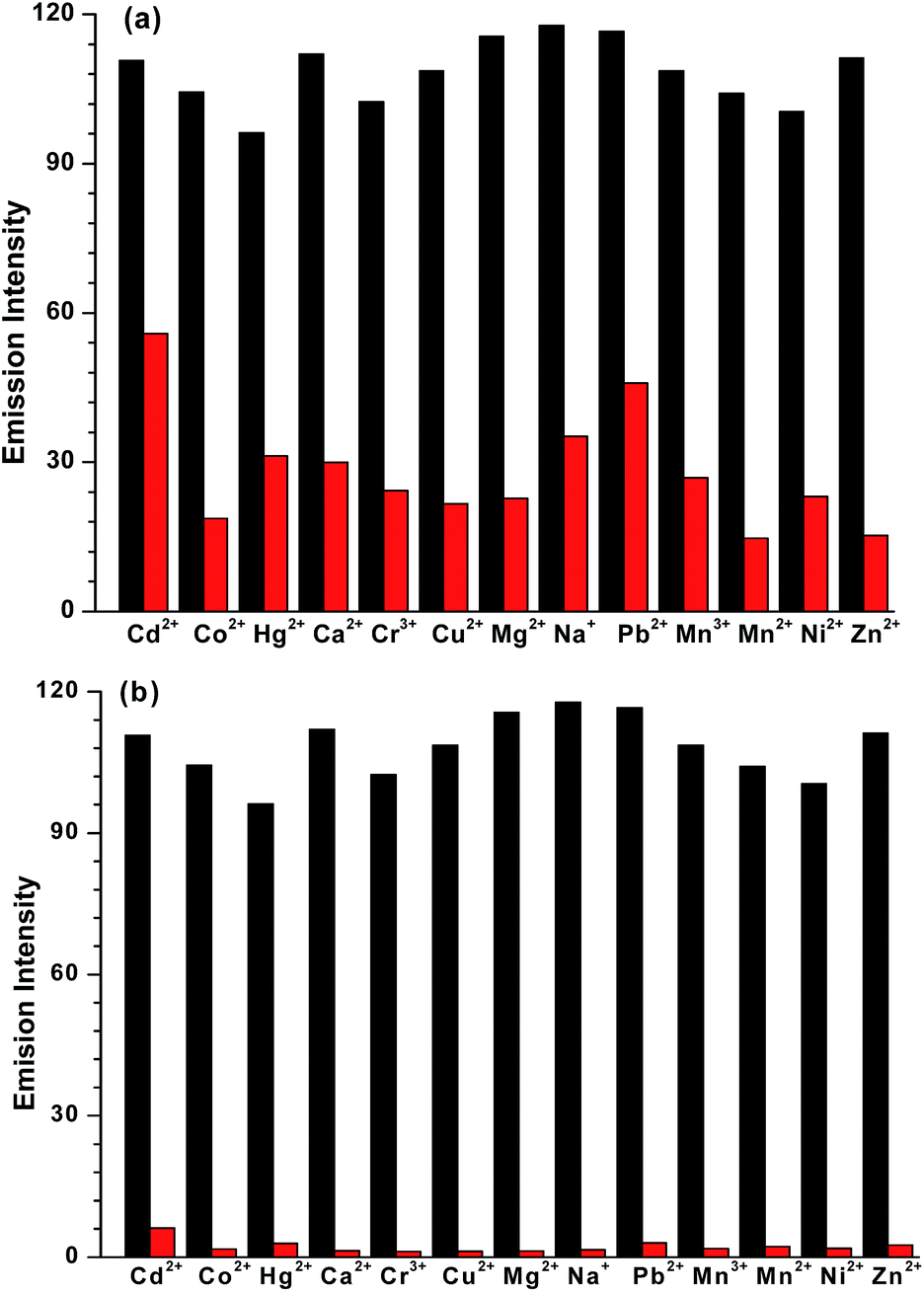
Competitive binding studies were performed to evaluate the interference from other metal ions such as Na+, Mg2+, Ca2+, Cr3+, Mn2+, Mn3+, Co2+, Ni2+, Cu2+, Zn2+, Hg2+, Cd2+ and Pb2+ in the detection of Fe2+ and Fe3+ ions. For the competitive binding studies, equal concentrations of either Fe2+ or Fe3+ ion and other metal ions were taken with L3 in THF. Fig. 4 displays that among the metal ions tested, no metal ion effectively interferes with the Fe2+ ion and especially with Fe3+ ion. Therefore, probe L3 acts as an effective sensor for Fe2+ and Fe3+ ions even in presence of other metal ions.
 |
| | Fig. 4 Selectivity of Fe2+ versus Fe3+ ion in a THF solution containing probe L3 + metal ions (black bar) and L3 + metal ions + (a) Fe2+ (red bar) or (b) Fe3+ (red bar) as observed by the fluorescence spectral studies. | |
Binding stoichiometry
The binding stoichiometry of anthracene-based probe L3 was investigated from Job's plot using fluorescence measurement.27 Emission intensity was plotted against mole fraction of Fe3+ ion at 440 nm (Fig. 5a). The maximum in Job's plot clearly indicates a 1:1 binding stoichiometry of Fe3+ ion with probe L3. The binding stoichiometry was also investigated by the Benesi–Hildebrand plot which further supported a 1:1 stoichiometry.28 An association constant, Ka, of 3.31 × 103 M−1 was determined by the ratio of intercept and slope in Benesi–Hildebrand plot (Fig. 5b). Notably, Benesi–Hildebrand plots either for 1:2 or 2:1 stoichiometries between L3 and Fe3+ ion showed poor fit therefore again confirming a 1:1 stoichiometry (Fig. S11 and S11b, ESI†).
 |
| | Fig. 5 (a) Job's plot of L3 and Fe3+ in THF. Total concentration of L3 and Fe3+ were maintained at 100 μM while the emission monitoring was done at 440 nm. (b) Benesi–Hildebrand plot for binding of Fe3+ ion with fluorescent probe L3. | |
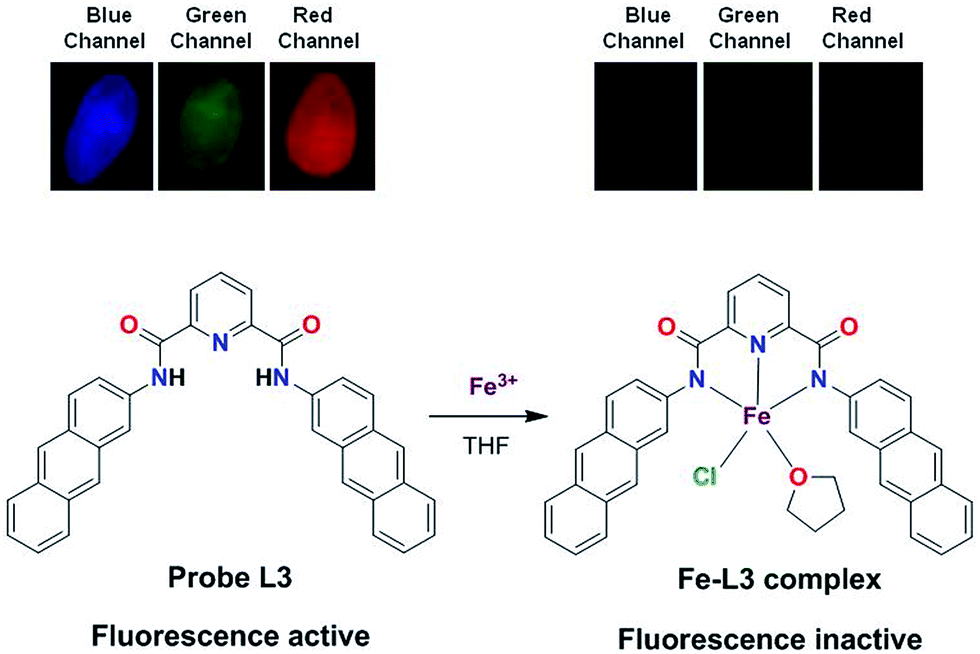
The Job's plot and Benesi–Hildebrand method suggested a 1:1 stoichiometry and formation of a Fe–L3 complex. To gain a clear understanding of the formulation of Fe–L3 complex, we isolated the product in a reaction of L3 and FeCl3 in THF (complex 1). This compound showed the disappearance of N–H stretches and bathochromic shift of Oamide frequency from 1657 (free L3) to 1602 cm−1 (Fig. S12, ESI†). Both these features suggest the coordination of Fe(III) ion by the deprotonated Namide groups within the pincer cavity.22d,29 To further identify the nature of this compound, high-resolution ESI+ mass spectrum was recorded which displays signals at m/z 701.4951 and at m/z 679.5123 corresponding to [L3 − 2H + Fe3+ + Cl + THF + Na+]+ and [L3 − 2H + Fe3+ + Cl + THF + H+]+, respectively (Fig. S13, ESI†). Therefore, mass spectrum corroborates the findings from the FTIR spectrum which suggest the involvement of deprotonated L3 in binding. Based on the collective studies, we propose a mononuclear formulation for the Fe–L3 complex (1) which is presented in Scheme 2.
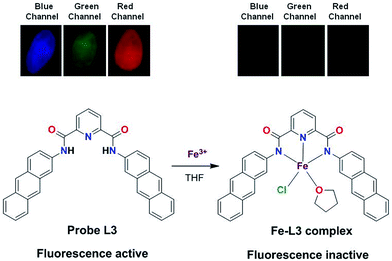 |
| | Scheme 2 Top: fluorescent images of solid sample of probe L3 and Fe–L3 complex in different channels (blue, green and red). Bottom: a proposed Fe–L3 complex formed by the 1:1 stoichiometry of Fe3+ ion and L3. | |
Interestingly, when complex 1 was recrystallized from DMF/diethyl ether, a new compound, complex 2, was obtained (Fig. S14, ESI†). Complex 2 was crystallographically characterized and the molecular structure is shown in Fig. 6. The Fe(III) ion is coordinated by four deprotonated Namide atoms in a tentative equatorial plane whereas two Npyridyl atoms occupy the axial positions. The Fe–Namide and Fe–Npy bond lengths are similar to a related Fe(III) complex reported by Mukherjee and co-workers.23 However, the Fe–Npy bond distances are larger than that observed for related structures.30 The five-membered chelate based Npyridine–Fe–Namide bond angles range from 81.4 to 82.6° whereas NPy–Fe–NPy bond angle is 179.5 Å. To confirm whether the same compound (2) can be synthesized independently by a 2:1 reaction between the deprotonated probe L3 and FeCl3 led to the isolation of 2 in high yield. We also recorded the emission spectrum of complex 2 and compared it either with a mixture of probe L3 and Fe3+ ion or isolated complex 1. As anticipated, complex 2 is fluorescence inactive, very similar to that of complex 1 or emission of L3 in presence of Fe3+ ion (Fig. S15, ESI†).
 |
| | Fig. 6 Ball and stick structure of the anionic part of complex 2; hydrogen atoms and tetrabutyl ammonium ion has been omitted for clarity. Selected bond distances (Å): Fe1–N1 = 1.8618, Fe1–N2 = 1.9415, Fe1–N3 = 1.9617. Selected bond angles (°): N1–Fe1–N2 = 82.6, N1–Fe1–N3 = 81.4, N2–Fe1–N3 = 164.0, N2–Fe1–N2# = 179.5. | |
Reversibility and logic gate applications
In order to develop a reversible probe for sensing Fe3+ ion, attempts were made to remove Fe3+ ion from the Fe–L3 complex. EDTA is known to have a strong tendency to chelate a variety of metals including Fe3+ ion and form a [Fe–EDTA]− complex.31 Fig. 7a displays that Fe3+ ion quenches the fluorescence intensity of probe L3 nearly quantitatively. However, addition of EDTA restores the fluorescence intensity of the probe L3 to nearly initial value.32 Further addition of Fe3+ ion again led to quantitative quenching. Such a fact establishes a reliable nature of probe L3. We have converted the sensing of Fe3+ ion in presence and absence of EDTA into logic gates which are used in several electronic devices such as molecular switches and keypads.33,34 We assumed that Fe3+ ion and EDTA are used as the inputs while the fluorescence intensity is used as an output. Emission intensity at 100 was taken as threshold frequency at 440 nm. Therefore, emission intensity above this threshold value gives OUT = 1 and below this OUT = 0. Consequently, these inputs (Fe3+ ion and EDTA) generate a truth table for OR gate as illustrated in Fig. 7b.
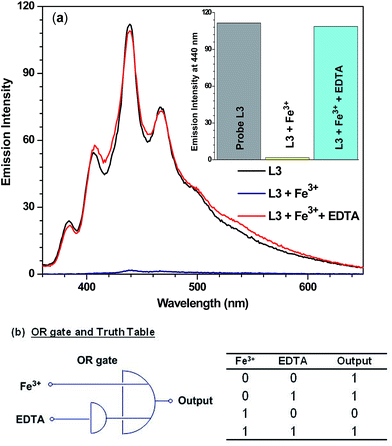 |
| | Fig. 7 (a) Emission spectra and bar diagram for change in emission intensity (inset) of L3 (40 μM) in presence of Fe3+ ion (10 equiv.) and EDTA (20 equiv.) in THF–water mixture.32 (b) Logic functions and truth table by change in emission intensity of L3 upon input of Fe3+ion and EDTA. | |
Cytotoxicity assays
As the probe L3 showed highest selectivity for Fe3+ ion out of three probes L1–L3; we selected it for the biological cell imaging applications.35 Before going to cell imaging, we attempted to ascertain the viability of cells towards probe L3 in absence and presence of Fe3+ ion. Therefore, cytotoxicity activity of L3 against HEK-293, Hep-2, and L929 cell lines was investigated in comparison with the widely used anti-cancer drug cis-platin under the identical conditions by using MTT assay.36 The in vitro cytotoxicity against HEK-293, Hep-2, and L929 cell lines is presented in Table 2. The anti-proliferative effects were tested using the MTT assay and were quantified in terms of IC50 values; lower the IC50 value, higher the anti-proliferative activity. The results show that probe L3 is less cytotoxic against all three HEK-293, Hep-2, and L929 cell lines as compared to probe L3 in presence of Fe3+ ion. Further, probe L3 in absence as well as in presence of Fe3+ ion showed significantly lower cytotoxicity as compared to cis-platin on all three tested cell lines. These studies advocate that the probe L3 can be used for the cell imaging in absence as well as presence of Fe3+ ion without causing any damage to the cell lines.
Table 2 IC50 values of in vitro cytotoxicity assay for HEK-293, Hep-2, and L929 cells
| Species |
IC50 (μM) |
| HEK-293 |
Hep-2 |
L929 |
| Probe L3 |
108.24 |
117.35 |
94.26 |
| Probe L3 + Fe3+ |
78.59 |
83.68 |
69.38 |
| cis-Platin |
13.38 |
12.62 |
11.05 |
Fluorescence cell imaging
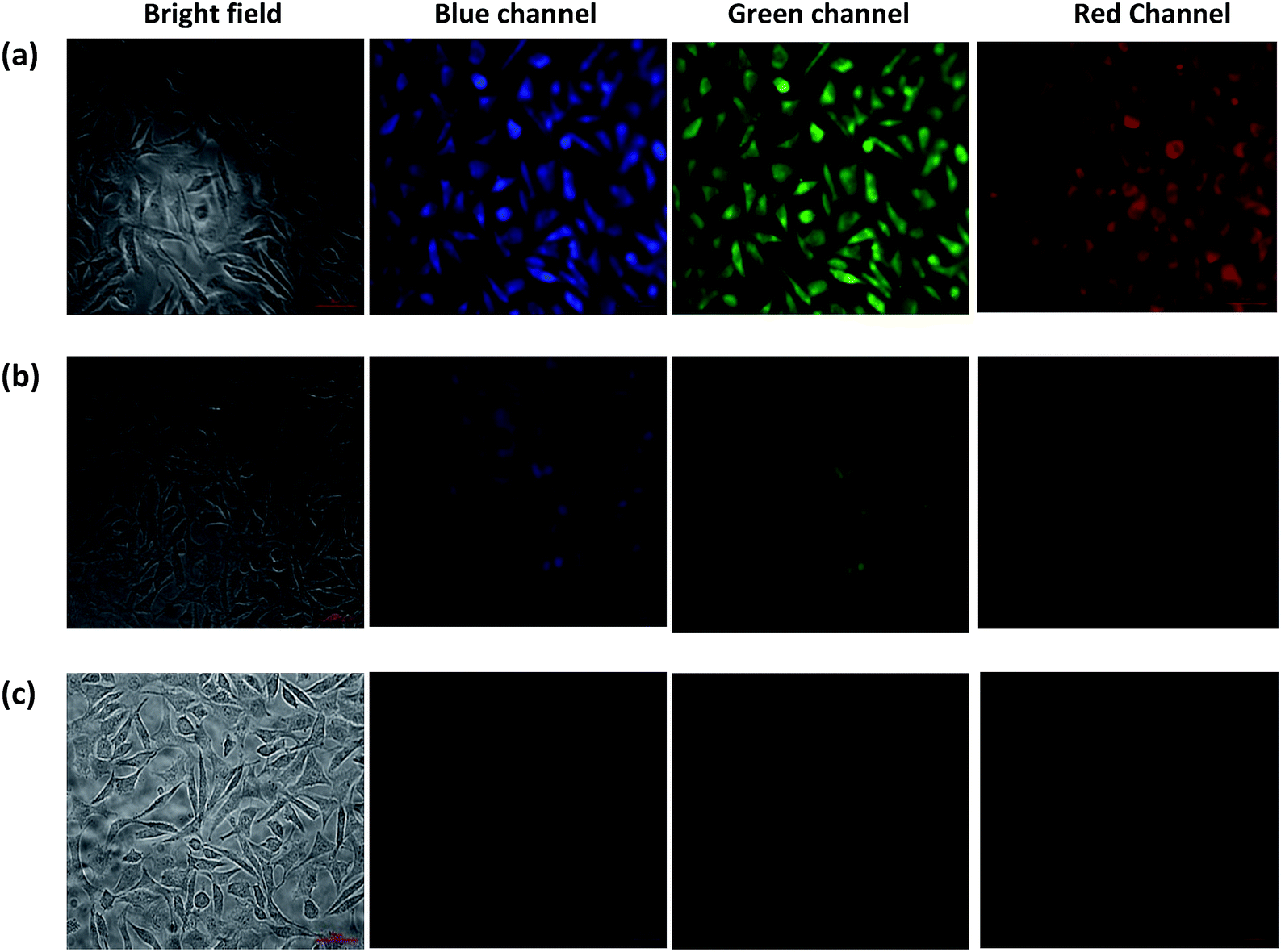
Cell cytotoxicity assay showed that the probe L3 has very low cytotoxicity; hence, can be used for the intracellular imaging. The application of probe L3 as an intracellular Fe3+ ion sensor was performed with L929 cells by fluorescence imaging. Fluorescence microscopic images of L929 cells for the blue, green and red channels are presented in Fig. 8. Examination of these images clearly illustrate that the probe L3 (40 μM) provides intense fluorescence inside the cells after its incubation for 90 min (Fig. 8a). Notably, incubation of probe L3 in presence of FeCl3 for 90 min, considerably quenches the fluorescence by the formation of Fe–L3 complex inside the cells (Fig. 8b). Further addition of FeCl3 quenches the fluorescence emission completely (Fig. 8c). We have already established that probe L3 coordinates with Fe3+ ion to form Fe–L3 complex which is fluorescent inactive. It can, therefore, be concluded that the probe L3 provides an effective mean to measure Fe3+ ion concentration within the living cells. Importantly, solid samples of probe L3 also display a similar quenching on exposure to ferric ion (cf. top portion of Scheme 2) thus suggesting wider applications.
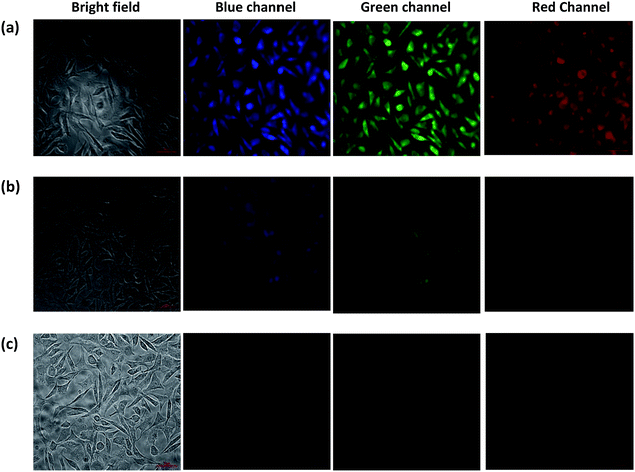 |
| | Fig. 8 Fluorescent microscopic images (40×) of L929 cells after incubation for 90 minutes in growth medium: (a) cells treated with probe L3 (40 μm); (b) cells treated with probe L3 (40 μM) and FeCl3 (400 μM); (c) cells treated with probe L3 (40 μm) and FeCl3 (1 mM). | |
Conclusions
In this report, a series of pincer-cavity based probes containing phenyl (L1), naphthyl (L2) and anthracenyl (L3) groups has been synthesized and studied. Probes L2 and L3 selectively detected iron both in +2 and +3 oxidation states. Notably, probe L3 afforded the best results when compared to L1 and L2. Fluorescence quenching, competitive binding studies, Stern–Volmer plots and detection limit advocated strong sensing ability of probe L3 for the Fe3+ ion as compared to Fe2+ ion. A 1:1 binding stoichiometry between L3 and Fe3+ ion was established with Job's plot with an association constant (Ka) of 3.31 × 103 M−1. Reversibility with EDTA demonstrated an intricate application in logic gate devices. Probe L3 was successfully used for the fluorescence based cell imaging and the results are supported with cell viability studies. Although iron selective probes have not been thoroughly investigated and optimized as compared to other metals; present results provide a strategy of developing pincer-cavity based selective probes and may find applications in tracking labile iron in living cells and tissues.
Experimental section
Materials and methods
All reagents and metal salts were used as received without further purifications. HPLC grade solvents were used for the UV-Vis and fluorescence measurements. Probes L1 and L2 were synthesised as per the literature reports.23,24
Syntheses of probe L3
Pyridine-2,6-dicarboxylic acid (1.0 g, 0.0059 mol) and 2-aminoanthracene (1.196 g, 0.0119 mol) were taken in 25 mL pyridine and refluxed with stirring for 30 min at 120 °C. P(OPh)3 (3.877 g, 0.0125 mol) was added drop-wise to the aforementioned reaction mixture. The reaction mixture was finally stirred at 80 °C for 12 h. After cooling to room temperature, the reaction mixture was poured into ice-cold water that caused precipitation of the product, which was filtered, washed with water followed by ethanol and dried in vacuum. Yield: 2.48 g (80%). Anal. calc. for C35H23N3O2: C, 81.22; H, 4.48; N, 8.12. Found: C, 81.24; H, 4.50; N, 8.15. FTIR spectrum (cm−1): 3270 (N–H), 1657 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). UV/Vis (THF): λmax (ε, M−1 cm−1) = 280 (74220), 310 (46200), 325 (42820), 375 (22120), 395 (15400). ESI+-MS spectrum (CH3CN, m/z): 518.1868 for [L3 + H+]. 1H NMR spectrum (400 MHz, DMSO-d6): δ = 11.31 (s, 2H), 8.81 (s, 2H), 8.59 (s, 4H),8.52–8.47 (d, J = 8.04 Hz, 2H), 8.40–8.34 (t, J = 8.56 Hz, 1H), 8.23–8.18 (d, J = 9.32 Hz, 2H), 8.11–8.06 (d, J = 7.92 Hz, 4H), 8.05–8.00 (d, J = 9.16 Hz, 2H) 7.57–7.45 (m, 4H). 13C NMR spectrum (100 MHz, DMSO-d6): 162.59 (C4), 149.40 (C3), 135.68 (C5), 132.28 (C1), 131.96 (C9), 131.31 (C13), 129.46 (C15), 129.39 (C16), 128.66 (C8), 128.36 (C14), 126.54 (C17), 126.32 (C2), 126.07 (C12), 126.03 (C10), 125.81 (C11), 122.77 (C6), 117.04 (C7).
O). UV/Vis (THF): λmax (ε, M−1 cm−1) = 280 (74220), 310 (46200), 325 (42820), 375 (22120), 395 (15400). ESI+-MS spectrum (CH3CN, m/z): 518.1868 for [L3 + H+]. 1H NMR spectrum (400 MHz, DMSO-d6): δ = 11.31 (s, 2H), 8.81 (s, 2H), 8.59 (s, 4H),8.52–8.47 (d, J = 8.04 Hz, 2H), 8.40–8.34 (t, J = 8.56 Hz, 1H), 8.23–8.18 (d, J = 9.32 Hz, 2H), 8.11–8.06 (d, J = 7.92 Hz, 4H), 8.05–8.00 (d, J = 9.16 Hz, 2H) 7.57–7.45 (m, 4H). 13C NMR spectrum (100 MHz, DMSO-d6): 162.59 (C4), 149.40 (C3), 135.68 (C5), 132.28 (C1), 131.96 (C9), 131.31 (C13), 129.46 (C15), 129.39 (C16), 128.66 (C8), 128.36 (C14), 126.54 (C17), 126.32 (C2), 126.07 (C12), 126.03 (C10), 125.81 (C11), 122.77 (C6), 117.04 (C7).
Synthesis of complex 1
Probe L3 (0.10 g, 0.193 mmol) was dissolved in THF (5 mL) followed by the drop-wise addition of a solution of FeCl3 (0.312 g, 1.93 mmol) dissolved in 5 mL THF. The reaction mixture was stirred for 2 h at room temperature. The solvent was removed under reduced pressure and the crude compound thus obtained was washed with diethyl ether and dried. FTIR spectrum (cm−1): 1602 (CO), 1454, 1346. UV/Vis spectrum (THF): λmax (ε, M−1 cm−1) = 330 (84270), 373 (46250), 395 (24900). ESI-MS spectrum (THF, m/z): 679.5123 for [L3 − 2H + Fe + Cl + THF + H+] and 701.4951 for [L3 − 2H + Fe + Cl + THF + Na+].
Synthesis of complex 2
Probe L3 (0.10 g, 0.193 mmol) was dissolved in THF (5 mL) and treated with solid NaH (0.092 g, 0.386 mmol). The resulting solution was stirred for 10 min at room temperature followed by the drop-wise addition of a solution of FeCl3 (0.031 g, 0.193 mmol) dissolved in 5 mL THF. After 15 min, tetrabutyl ammonium bromide (0.062 g, 0.193 mmol) was added and the reaction mixture was stirred for 2 h at room temperature. The solvent was removed under the reduced pressure and the crude compound thus obtained was washed with diethyl ether and dried. Reddish brown crystals were obtained by the vapour diffusion of diethyl ether into a DMF solution of compound within one week. Yield: 0.16 g (74%). Anal. calc. for C70H42N6O4FeNa: C, 75.75; H, 3.81; N, 7.57. Found: C, 75.72; H, 3.83; N, 7.54. FTIR spectrum (cm−1): 1601 (CO), 445(7460). 1456, 1347. UV/Vis (THF): λmax (ε, M−1 cm−1) = 375 (21560), 395 (17020).
Physical methods
Elemental analysis data were obtained by Elementar Analysen Systeme GmbH Vario EL-III instrument. The 1H and 13C NMR spectra were recorded with a JEOL 400 MHz instrument. The FTIR spectra (Zn–Se ATR) were recorded with a Perkin-Elmer Spectrum-Two spectrometer. The absorption spectra were recorded with a Perkin-Elmer Lambda 25 spectrophotometer. Fluorescence studies were performed with a Cary Eclipse Fluorescence Spectrometer. ESI-MS mass spectra were obtained with Q-TOF LC/MS mass spectrometer of Agilent.
UV-Visible and fluorescence measurements
All stock solutions of L1, L2 and L3 (1 mM) and metal salts (10 mM) of Na+, Mg2+, Ca2+, Cr3+, Mn2+, Mn3+, Fe2+, Fe3+, Co2+, Ni2+, Cu2+, Zn2+, Hg2+, Cd2+ and Pb2+ ions were prepared in THF. All UV-Vis and fluorescence spectra were recorded with a 1.0 cm path length cuvette.
Determination of Stern–Volmer constant (KSV) and detection limit
Fluorescence titrations were further evaluated using the Stern–Volmer equation [eqn (1)].25where I0 is the emission intensity in the absence of quencher, I is the emission intensity in the presence of quencher (Q) and KSV is the Stern–Volmer constant.
The detection limit was calculated using eqn (2),
| | |
Detection limit: 3σ/k
| (2) |
where
σ is the standard deviation of a blank measurements and
k is the slope of a plot of fluorescence intensity
versus metal ion concentration.
26
Determination of the binding stoichiometry and the apparent association constant Ka
The binding stoichiometry of Fe3+ with L3 was determined by the Job's plot.27 The fluorescence intensity at 440 nm was plotted against mole fraction of probe L3 at constant concentration. The apparent association constant (Ka) of Fe–L3 complexes was determined by the Benesi–Hildebrand equation.28 The association constant Ka was calculated graphically by plotting 1/(I − I0) against 1/[Fe3+].| | |
1/(I − I0) = 1/{Ka(I0 − Imin)[Fe3+]} + 1/(I0 − Imin)
| (3) |
where I is the fluorescence intensity of L3 in presence of Fe(III) ion at 440 nm, I0 is the intensity of L3 in absence of Fe3+ ion and Imin is the minimum fluorescence intensity in presence of Fe3+ ion. The plot 1/(I − I0) vs. 1/[Fe3+] were linearly fitted and the Ka value was obtained from the slope and intercept of the line.
In vitro cytotoxicity assay
The cytotoxicity potential of the fluorescent probe L3 in absence and presence of Fe3+ was evaluated by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay against HEK-293, Hep-2, and L929 cells.36 HEK-293, Hep-2, and L929 cells (1 × 104) were seeded on 96-well plates in growth medium and incubated at 37 °C in an atmosphere containing 5% CO2. After incubation of 24 h, testing solutions of serial concentrations were added to each well and incubated for next 24 h. The tested solutions were made in DMEM medium with DMSO for better solubility while the final concentration of DMSO was 1%. At the end of each treatment, cell viability was assessed according to the manufacturer's instructions. Briefly, after discarding the medium, MTT labelling mixture (5 mg mL−1) was added to the cells and incubated for 2 h at 37 °C. The media with MTT were removed and 100 μM of DMSO was added to dissolve the insoluble formazon. Absorbance values were determined on Fluostar optima (BMG Labtech, Germany) microplate reader at 570 nm. The percentage of inhibition was calculated as:
The IC50 (50% inhibitory concentration) values were obtained by the nonlinear regression, using the Graph Pad Prism version 5 program. All experiments were independently repeated at least three times, with triplicate samples for each treatment.
Fluorescence cell imaging
In vitro studies with probe L3 in presence as well as in absence of Fe3+ were carried out with L929 cells (murine cancer cells). L929 cells were cultured in DMEM medium supplemented with 1% streptomycin as antibiotic, and incubated in CO2 incubator at 37 °C. Cells were seeded at a density of 2.0 × 105 cells per well in six well plate and were incubated at 37 °C for 12 h. Cells were incubated with probe L3 (40 μM) in phosphate buffer saline (PBS) for 90 min. In another set of experiment, cells were incubated with probe L3 (40 μM) and FeCl3 (400 μM and 1 mM). After 90 min incubation, cells were washed with PBS three times to remove the traces of probe L3. Finally the cells were fixed with 4% paraformaldehyde solution. Stained slides were observed and acquired the image in Upright Microscope (Axio Imager 2.2, Carl Zeiss, Germany).
X-ray crystallography
Single crystals suitable for the X-ray diffraction studies were grown by the vapor diffusion of diethyl ether into a dimethyl formamide solution of complex 2. The intensity data were collected at 298 K with an Oxford XCalibur CCD diffractometer equipped with graphite monochromatic Mo-Kα radiation (λ = 0.71073 Å).37 Data reduction was performed with the CrysAllisPro program (Oxford Diffraction ver. 171.34.40). The structure was solved using the direct methods using SHELXS-97 (ref. 38) program and refined on F2 using all data by full matrix least-squares procedures with SHELXL-97 (ref. 39) within the OLEX-2 suite.40 The hydrogen atoms were placed at the calculated positions and included in the last cycles of the refinement. All calculations were done using the WinGX software package.41 Crystallographic data collection and structure solution parameters are summarized in Table S1 (ESI).†
Acknowledgements
RG acknowledges the Council of Scientific and Industrial Research (CSIR), New Delhi and the University of Delhi for the financial support. PK thanks UGC, New Delhi for the Dr D. S. Kothari Postdoctoral fellowship. Authors thank the CIF-USIC at this university for the instrumental facilities and crystallographic data collection and Dr Arun Kumar (IIT Delhi) for technical assistance in cell imaging work.
Notes and references
-
(a) K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS PubMed;
(b) X. Chen, T. Pradhan, F. Wang, J. S. Kim and J. Yoon, Chem. Rev., 2012, 112, 1910–1956 CrossRef CAS PubMed;
(c) M. E. Jun, B. Roy and K. H. Ahn, Chem. Commun., 2011, 47, 7583–7601 RSC;
(d) J. Du, M. Hu, J. Fan and X. Peng, Chem. Soc. Rev., 2012, 41, 4511–4535 RSC.
-
(a) H. N. Kim, W. X. Ren, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2012, 41, 3210–3244 RSC;
(b) T. Nagano, Y. Gabe, Y. Urano, K. Kikuchi and H. Kojima, J. Am. Chem. Soc., 2004, 126, 3357–3367 CrossRef PubMed;
(c) T. Terai, Y. Urano, S. Izumi, H. Kojima and T. Nagano, Chem. Commun., 2012, 48, 2840–2842 RSC.
- R. C. Hider and X. Kong, Dalton Trans., 2013, 42, 3220–3229 RSC.
-
(a) E. Beutler, Science, 2004, 306, 2051–2053 CrossRef CAS PubMed;
(b) P. L. Llewellyn, P. Horcajada, G. Maurin, T. Devic, N. Rosenbach, S. Bourrelly, C. Serre, D. Vincent, S. Loera-Serna, Y. Filinchuk and G. Ferey, J. Am. Chem. Soc., 2009, 131, 13002–13008 CrossRef CAS PubMed.
-
(a) S. Dai, C. Schwendtmayer, P. Schurmann, S. Ramaswamy and H. Eklund, Science, 2000, 287, 655–658 CrossRef CAS PubMed;
(b) A. Atkinson and D. R. Winge, Chem. Rev., 2009, 109, 4708–4721 CrossRef CAS PubMed;
(c) C. D. Kaplan and J. Kaplan, Chem. Rev., 2009, 109, 4536–4552 CrossRef CAS PubMed;
(d) E. C. Theil and D. J. Goss, Chem. Rev., 2009, 109, 4568–4579 CrossRef CAS PubMed.
-
(a) D. Galaris, V. Skiada and A. Barbouti, Cancer Lett., 2008, 266, 21–29 CrossRef CAS PubMed;
(b) N. K. Desai, G. B. Kolekar and S. R. Patil, New J. Chem., 2014, 38, 4394–4403 RSC.
-
(a) B. Halliwell, J. Neurochem., 1992, 59, 1609–1623 CrossRef CAS PubMed;
(b) E. D. Weinberg, Eur. J. Cancer Prev., 1996, 5, 19–36 CAS.
- P. Das, N. B. Chandar, S. Chourey, H. Agarwalla, B. Ganguly and A. Das, Inorg. Chem., 2013, 52, 11034–11041 CrossRef CAS PubMed.
-
(a) C. X. Yang, H. B. Ren and X. P. Yan, Anal. Chem., 2013, 85, 7441–7446 CrossRef CAS PubMed;
(b) J. Brandel, N. Humbert, M. Elhabiri, I. J. Schalk, G. L. Mislin and A. M. Albrecht-Gary, Dalton Trans., 2012, 41, 2820–2834 RSC.
-
(a) Y. Ma, W. Luo, P. J. Quinn, Z. Liu and R. C. Hider, J. Med. Chem., 2004, 47, 6349–6362 CrossRef CAS PubMed;
(b) X. Qu, Q. Liu, X. Ji, H. Chen, Z. Zhou and Z. Shen, Chem. Commun., 2012, 48, 4600–4602 RSC;
(c) Q. X. Yao, J. L. Sun, K. Li, J. Su, M. V. Peskov and X. Zou, Dalton Trans., 2012, 41, 3953–3955 RSC.
-
(a) B. Wang, J. Hai, Z. Liu, Q. Wang, Z. Yang and S. Sun, Angew. Chem., Int. Ed., 2010, 49, 4576–4579 CrossRef CAS PubMed;
(b) S. K. Sahoo, D. Sharma, R. K. Bera, G. Crisponi and J. F. Callan, Chem. Soc. Rev., 2012, 41, 7195–7227 RSC.
-
(a) B. Bodenant, F. Fages and M.-H. Delville, J. Am. Chem. Soc., 1998, 120, 7511–7519 CrossRef CAS;
(b) M. Xu, S. Wu, F. Zeng and C. Yu, Langmuir, 2010, 26, 4529–4534 CrossRef CAS.
-
(a) J. Mao, L. Wang, W. Dou, X. Tang, Y. Yan and W. Liu, Org. Lett., 2007, 9, 4567–4570 CrossRef CAS;
(b) Y. Xiang and A. Tong, Org. Lett., 2006, 8, 1549–1552 CrossRef CAS PubMed.
- S. Fakih, M. Podinovskaia, X. Kong, U. E. Schaible, H. L. Collins and R. C. Hider, J. Pharm. Sci., 2009, 98, 2212–2226 CrossRef CAS PubMed.
-
(a) H. Weizman, O. Ardon, B. Mester, J. Libman, O. Dwir, Y. Hadar and Y. Chen, J. Am. Chem. Soc., 1996, 118, 12368–12375 CrossRef CAS;
(b) W. D. Chen, W. T. Gong, Z. Q. Ye, Y. Lin and G. L. Ning, Dalton Trans., 2013, 42, 10093–10096 RSC;
(c) M. H. Lee, T. V. Giap, S. H. Kim, Y. H. Lee, C. Kang and J. S. Kim, Chem. Commun., 2010, 46, 1407–1409 RSC.
-
(a) G. Mukherjee and K. Biradha, Chem. Commun., 2012, 48, 4293–4295 RSC;
(b) T. K. Prasad, D. H. Hong and M. P. Suh, Chem.–Eur. J., 2010, 16, 14043–14050 CrossRef CAS PubMed;
(c) L. W. Mi, H. W. Hou, Z. Y. Song, H. Y. Han and Y. T. Fan, Chem.–Eur. J., 2008, 14, 1814–1821 CrossRef CAS PubMed.
- Z. Yang, M. She, B. Yin, J. Cui, Y. Zhang, W. Sun, J. Li and Z. J. Shi, J.Org. Chem., 2012, 77, 1143–1147 CrossRef CAS PubMed.
-
(a) S. K. Sahoo, D. Sharma, R. K. Bera, G. Crisponi and J. F. Callan, Chem. Soc. Rev., 2012, 41, 7195–7227 RSC;
(b) H. N. Kim, M. H. Lee, H. J. Kim, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2008, 37, 1465–1472 RSC.
- A. Dorazco-Gonzalez, M. F. Alamo, C. Godoy-Alcantar, H. Hopfl and A. K. Yatsimirsky, RSC Adv., 2014, 4, 455–466 RSC.
- A. Dorazco-Gonzalez, H. Hopfl, F. Medrano and A. K. Yatsimirsky, J. Org. Chem., 2010, 75, 2259–2273 CrossRef CAS PubMed.
-
(a) P. J. Donoghue, A. K. Gupta, D. W. Boyce, C. J. Cramer and W. B. Tolman, J. Am. Chem. Soc., 2010, 132, 15869–15871 CrossRef CAS PubMed;
(b) P. Pirovano, A. M. Magherusan, C. McGlynn, A. Ure, A. Lynes and A. R. McDonald, Angew. Chem., Int. Ed., 2014, 53, 5946–5950 CrossRef CAS PubMed;
(c) P. J. Donoghue, J. Tehranchi, C. J. Cramer, R. Sarangi, E. I. Solomon and W. B. Tolman, J. Am. Chem. Soc., 2011, 133, 17602–17605 CrossRef CAS PubMed;
(d) A. K. Patra and R. Mukherjee, Inorg. Chem., 1999, 38, 1388–1393 CrossRef CAS;
(e) A. K. Singh, V. Balamurugan and R. Mukherjee, Inorg. Chem., 2003, 42, 6497–6502 CrossRef CAS PubMed.
-
(a) D. Huanga, O. V. Makhlynetsb, L. L. Tan, S. C. Lee, E. V. Rybak-Akimova and R. H. Holm, Proc. Nat. Acad. Sci., 2011, 108, 1222–1227 CrossRef PubMed;
(b) X. Zhang, D. Huang, Y.-S. Chen and R. H. Holm, Inorg. Chem., 2012, 51, 11017–11029 CrossRef CAS PubMed;
(c) D. Huang and R. H. Holm, J. Am. Chem. Soc., 2010, 132, 4693–4701 CrossRef CAS PubMed;
(d) D. Bansal, G. Kumar, G. Hundal and R. Gupta, Dalton Trans., 2014, 43, 14865–14875 RSC.
- M. Ray, D. Ghosh, Z. Shirin and R. Mukherjee, Inorg. Chem., 1997, 36, 3568–3572 CrossRef CAS PubMed.
- K. Ghosh, S. Kumar, R. Kumar and U. P. Singh, J. Organomet. Chem., 2014, 750, 169–175 CrossRef CAS.
- A. Ganguly, B. K. Paul, S. Ghosh, S. Kar and N. Guchhait, Analyst, 2013, 138, 6532–6541 RSC.
- C. Liang, W. Bu, C. Li, G. Men, M. Deng, Y. Jiangyao, H. Sun and S. Jiang, Dalton Trans., 2015, 42, 1827–1833 Search PubMed.
- A. Senthilvelan, I. Ho, K. Chang, G. Lee, Y. Liu and W. Chung, Chem.–Eur. J., 2009, 15, 6152–6160 CrossRef CAS PubMed.
-
(a) H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS;
(b) R. B. Singh, S. Mahanta and N. Guchhait, J. Mol. Struct., 2010, 963, 92–97 CrossRef CAS.
- S. Srivastava, M. S. Dagur and R. Gupta, Eur. J. Inorg. Chem., 2014, 4966–4974 CrossRef CAS.
- D. S. Marlin, M. M. Olmstead and P. K. Mascharak, Inorg. Chem., 1999, 38, 3258–3260 CrossRef CAS.
- E. M. A. Zaitoun and C. T. Lin, J. Phys. Chem. B, 1997, 101, 1857–1860 CrossRef.
- EDTA was added to a THF–water (40%, v/v) solution of probe L3 and Fe3+ ion. Under these conditions, emission spectrum of probe L3 and its interaction with Fe3+ ion was similar as noted in neat THF.
-
(a) M. Kumar, A. Dhir and V. Bhalla, Org. Lett., 2009, 11, 2567–2570 CrossRef CAS PubMed;
(b) V. Bhalla, Roopa and M. Kumar, Org. Lett., 2012, 14, 2802–2805 CrossRef CAS PubMed.
-
(a) D. Margulies, C. E. Felder, G. Melman and A. Shanzer, J. Am. Chem. Soc., 2007, 129, 347–354 CrossRef CAS PubMed;
(b) B. Rout, P. Milko, M. A. Iron, L. Motiei and D. Margulies, J. Am. Chem. Soc., 2013, 135, 15330–15333 CrossRef CAS PubMed.
- For cell imaging, 1% DMSO–water (v/v) mixture and DMEM buffer was used. Under these conditions, the emission spectrum of probe L3 and its interaction with Fe3+ ion was similar as noted in neat THF.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- CrysAlisPro, v. 1.171.33.49b, Oxford Diffraction Ltd., 2009.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXL-97, Programs for X-ray Crystal Structure Refinement, University of Göttingen, Gottingen, Germany, 1997 Search PubMed.
- O. V. Dolomanov, L. J. Bouhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Figures for the FTIR, UV-Vis, ESI-MS and NMR spectral studies, fluorescence spectral titration studies, detection limits; and a crystallographic table. CCDC 1424233. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra20760f |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.