
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
11C-carbonylation reactions using gas–liquid segmented microfluidics†
Kenneth
Dahl
*a,
Magnus
Schou
b,
Johan
Ulin
c,
Carl-Olof
Sjöberg
c,
Lars
Farde
ab and
Christer
Halldin
a
aKarolinska Institutet, Department of Clinical Neuroscience, Centre for Psychiatric Research, Karolinska Hospital, S-171 76 Stockholm, Sweden. E-mail: Kenneth.dahl@ki.se
bAstraZeneca Translational Science Centre, Department of Clinical Neuroscience, Karolinska Institutet, S-171 76 Stockholm, Sweden
cBencar AB, Uppsala Science Park, S-751 83 Uppsala, Sweden
First published on 20th October 2015
Abstract
A novel gas–liquid segmented microfluidic platform has been developed. The Pd-mediated 11C-carbonylation reaction proceeds smoothly on this platform and good to excellent radiochemical conversions (RCC) were observed. Twelve compounds were successfully radiolabelled using this novel technology, including the well established D2 receptor radioligands [11C]raclopride and [11C]FLB 457.
[11C]Carbon monoxide (11CO), derived from the positron emitting nuclide 11C (t1/2 = 20.4 min), is an attractive synthon in PET (Positron Emission Tomography) radiochemistry,1 as the carbonyl group is present in most biologically-relevant molecules. Consequently, a great deal of research efforts has been devoted to developing efficient and simple methods for its introduction, e.g. high-pressure reactors,2 xenon gas carrier,311CO trapping solutions,4 reactive catalytic species,5 oxidant reagents6 and backed-tube reactors.7 In our long-term objective to improve general access to this synthon, we turned our attention to microfluidic (MF) technology with its well documented advantages over conventional batch reactions.8 In particular, multi-phase MF, which offers advantages such as large interfacial areas, fast mixing, precision temperature control and reduced mass-transfer limitations. Two distinctly different flow conditions exist for gas–liquid MF reactions. The first condition is commonly referred to as annular flow and is characterized by a gas flow in the centre of a liquid film coated on the internal surface of the reactor. The second flow condition is called segmented flow and relies on the continuous formation of micro-bubbles within the liquid flow. In general, the segmented flow approach provides better control over reaction condition and more importantly have proven to reduce the formation of Pd particles, which clog the MF channel.9
MF is a rapidly growing field within PET radiochemistry,10 however, until this day, its application in 11C-radiochemistry remain rather unexplored. In 2004, Lu et al. reported the first 11C-synthesis using a MF approach.11 A glass fabricated, T-shaped micro reactor was used to study the liquid–liquid MF reaction of carboxylic acids with [11C]methyl iodide as methylating agent. More recently, Miller et al. presented a Pd-mediated carbonylative protocol to 11C-labelled products, using a gas–liquid MF approach.12 The heterogeneous reaction was performed by generating an annular flow of 11CO/N2 inside a 5 m long serpentine-shaped micro channel, prefilled with coupling reagent solution. Later, a commercially available MF device was used to perform liquid–liquid phase 11C-carbonylation reactions, in which a liquid solution of Cu(Tp*)11CO was applied as CO donor.13 The system was applied in the synthesis of the neuropeptide Y5 receptor antagonist, [11C]MK-9233.14
In this communication we report the first application of a gas–liquid segmented MF protocol allowing direct access to an array of 11C-labelled drug-like amides. In addition to the labeling of [11C]amides, the protocol also demonstrated its utility in the radiosynthesis of a [11C]carboxylic acid and three [11C]esters.
The MF system (Fig. 1) used in this study consists of a precision syringe pump, a μ-mass flow controller, a mixing-tee to permit gas-to-liquid contact, and a 5 m fused-silica capillary reactor (inner diameter (i.d.) = 200 μm) located within a preheated oil bath, as well as a back-pressure regulator (100 psi, BPR). In a typical reaction, 11CO was trapped and concentrated on a small silica column at −196 °C. The accumulated 11CO was subsequently transferred into the MF reactor using the μ-mass flow controller charged with helium as carrier. At the same time a premixed solution of coupling reagents (aryl halide, Pd-ligand and amine in anhydrous THF) was infused into the MF reactor using the syringe pump. A leak-tight gas bag was connected to the outlet of the product vial to receive volatile radioactive products (e.g.11CO). The fully automated synthesis process was controlled and monitored using in-house developed software (for full experimental details see the ESI†).
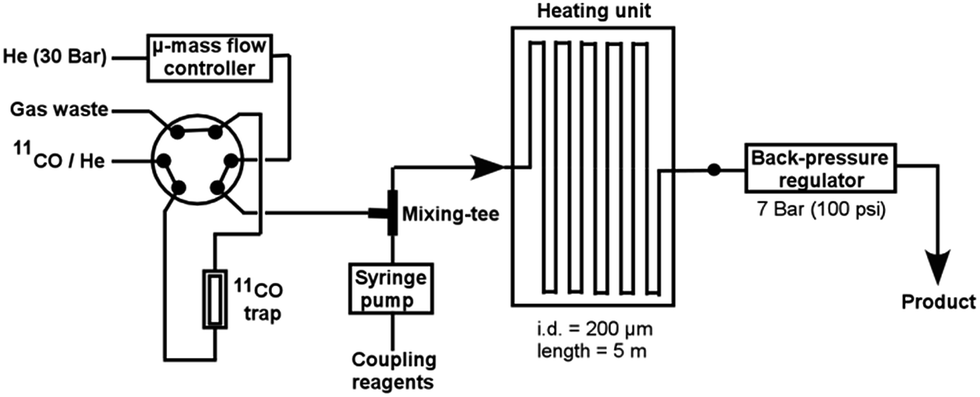 | ||
| Fig. 1 Schematic diagram of the microfluidic system. | ||
Initially, experiments were performed at different flow rates using a micro mixing-tee (i.d. = 50 μm) in order to identify conditions with sufficient gas-to-liquid interfacial area. Thus, a series of experiments was performed using the synthesis of N-benzyl-[carbonyl-11C]benzamide ([11C]3) as a model reaction using Pd(PPh3)4 as catalyst. As expected, the RCC of [11C]3 was strongly dependent on the gas-to-liquid flow rates. For example, by decreasing the gas flow from 200 μL min−1 to 100 μL min−1 while keeping the liquid flow constant (20 μL min−1), a close to 3-fold improvement in RCC was observed (Table 1, entries 1 and 2). Next we examined the reaction at different temperatures. No notable improvement was observed at 120 °C (Table 1, entry 3) compared to 100 °C. Attempts to perform the reaction at lower temperatures resulted in decreased 11CO trapping efficiency and thereby lower RCC (Table 1, entry 4). On the other hand, a quantitative conversion to the desired product was observed already at room temperature (r.t.) using Pd2(cinnamyl)Cl2–xantphos as catalyst (Table 1, entry 6). This further illustrates the utility of Pd2(cinnamyl)Cl2–xantphos in 11C-aminocarbonylation reactions.5 During the course of the condition screening, we experienced issues related to clogging of the micro mixing-tee. In order to improve the robustness of the method, we decided to test a mixing-tee with a larger inner diameter (i.d. = 150 μm). Further alterations to the conditions were thus conducted (Table 1, entries 7–9). To our delight, at 100 °C, a gas flow of 100 μL min−1, liquid flow of 30 μL min−1 using Pd(PPh3)4 as catalyst, [11C]3 was obtained in a reproducible RCC of 95 ± 1% (Table 1, entry 8).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entrya | T (°C) | Gas flow (μL min−1) | Liquid flow (μL min−1) | Mixing tee (i.d., μm) | Catalyst | Trapped 11COb (%) | RCPc (%) | RCCd (%) |
| a Reaction conditions: iodobenzene (20 μmol), benzylamine (50 μL), Pd-source (14 μmol), ligand (14 μmol), THF (1 mL), 100 °C. b Decay corrected; the fraction of radioactivity left in the crude product after purging with nitrogen. c Radiochemical purity determined by radioanalytical HPLC. d Radiochemical conversion based on the total radioactivity delivered to the collection vial. e Average of two runs. | ||||||||
| 1 | 100 | 200 | 20 | 50 | Pd(PPh3)4 | 53 | 71 | 37 |
| 2 | 100 | 100 | 20 | 50 | Pd(PPh3)4 | >99 | 96 | 95 ± 2e |
| 3 | 80 | 100 | 20 | 50 | Pd(PPh3)4 | 89 | 67 | 59 |
| 4 | 120 | 100 | 20 | 50 | Pd(PPh3)4 | >99 | 94 | 93 |
| 5 | 100 | 100 | 20 | 50 | Pd2(cinnamyl)Cl2–xantphos | >99 | 99 | 99 |
| 6 | r.t. | 100 | 20 | 50 | Pd2(cinnamyl)Cl2–xantphos | >99 | 98 | 98 |
| 7 | 100 | 100 | 20 | 150 | Pd(PPh3)4 | 95 | 91 | 86 |
| 8 | 100 | 100 | 30 | 150 | Pd(PPh3)4 | >99 | 96 | 95 ± 1e |
| 9 | 100 | 200 | 30 | 150 | Pd(PPh3)4 | 91 | 90 | 82 |

Furthermore, in order to explore the applicability of the developed method, the best conditions (Table 1, entries 5 and 8) were first applied in synthesis of a variety of 11C-labelled test compounds (Scheme 1, compound [11C]3–7). All reactions showed high 11CO trapping efficiency (>95%) and the test compounds were produced in a RCC range of 79–99%.
 | ||
| Scheme 1 Compounds produced using the gas–liquid segment microfluidic approach. Conditions A: aryl-halide, nucleophile, Pd(PPh3)4, THF, 100 °C. Conditions B: aryl-halide, nucleophile, Pd2(cinnamyl)Cl2, xantphos, THF, 100 °C. Conditions C: iodobenzene, benzylamine, [PdCl2–(xantphos)], toluene, 100 °C. Average of two runs. | ||
Finally, a series of drug-like amides were successfully radiolabelled using the methodology (Scheme 1, compound [11C]8–[11C]14). In general, good RCCs were observed when using Pd(PPh3)4 as catalyst, as exemplified by the well established D2 receptor radioligand, [11C]FLB 457 15 ([11C]8), which was produced in a RCC of 61 ± 4% with a near quantitative 11CO trapping efficiency. However, for [11C]13 and [11C]raclopride16 ([11C]14) Pd(PPh3)4 was found ineffective as a catalyst. For these molecules, the more active Pd2(cinnamyl)Cl2–xantphos catalytic system provided RCCs of 41 ± 1% and 79 ± 1%, respectively. The present MF platform has now been operated conveniently over 100 times without any experiences with clogging. When comparing the synthesis of [11C]13 in the current work with the previously reported gas–liquid annular MF approach,12 we observe a 12% increase in RCC with our setup. We attribute this finding to the larger gas–liquid interface generated using the gas–liquid segmented approach. An enlarged photo of the fused-silica capillary is shown in Fig. 2, in which this flow profile is confirmed.
 | ||
| Fig. 2 Photographic image of the flow profile inside the fused-silica capillary. | ||
PET radioligands for in vivo human use are typically produced in gigabecquerel (GBq) quantities, therefore, as a final statement to the utility of this method, two compound ([11C]12, 13) were produced on a preparative scale. Production data are summarized Table 2. All compounds were produced in sufficient radioactivity amounts (1200 and 2800 MBq), and with high radiochemical purity (RCP, >99%) and moderate specific radioactivity (SRA, 40 and 54 GBq μmol−1).
| Product | Isolated yield (MBq) | SRA (GBq μmol−1) | RCP (%) | Synthesis time (min) |
|---|---|---|---|---|

|
1200 | 40 | >99 | 49 |
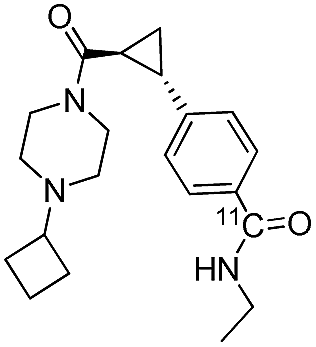
|
2800 | 54 | >99 | 52 |
Conclusions
In summary, a novel gas–liquid segmented microfluidic approach to the synthesis of 11C-carbonyl labelled compounds has been developed. To our knowledge this represents the first application of gas–liquid segmented microfluidics within the field of PET radiochemistry. The suitability of this technique was demonstrated with the synthesis of twelve different 11C-labelled compounds, including the well established D2 receptor radioligands [11C]raclopride and [11C]FLB 457.Acknowledgements
The authors would like to give a special thanks to Dr Peter Johnström and Dr Vladimir Stepanov. We also thank all members of the PET group at Karolinska Institutet for all their support. The work was conducted with financial support from StratNeuro and AstraZeneca.Notes and references
- (a) B. Långström, O. Itsenko and O. Rahman, J. Labelled Compd. Radiopharm., 2007, 50, 794 CrossRef; (b) S. Kealey, A. Gee and P. W. Miller, J. Labelled Compd. Radiopharm., 2014, 57, 195 CrossRef CAS PubMed; (c) O. Rahman, J. Labelled Compd. Radiopharm., 2015, 58, 86 CrossRef CAS PubMed.
- (a) T. Kihlberg and B. Långström, J. Org. Chem., 1999, 64, 9201 CrossRef CAS; (b) H. Burn and E. Hostetler, Nucl. Med. Biol., 2002, 29, 845 CrossRef; (c) T. Kihlberg and B. Långström, PCT Int. Appl., WO2002/102711 A1, 2002; (d) K. Dahl, O. Itsenko, O. Rahman, J. Ulin, C. Sjöberg, P. Sandblom, L. Larsson and C. Halldin, J. Labelled Compd. Radiopharm., 2015, 58, 220 CrossRef CAS PubMed.
- J. Eriksson, J. van den Hoek and A. D. Windhorst, J. Labelled Compd. Radiopharm., 2012, 55, 223 CrossRef CAS.
- (a) H. Audrain, L. Martarello, A. Gee and D. Bender, Chem. Commun., 2004, 558 RSC; (b) S. Kealey, P. W. Miller, N. Long, C. Pilsson, L. Martarello and A. Gee, Chem. Commun., 2009, 3696 RSC.
- (a) K. Dahl, M. Schou, N. Amini and C. Halldin, Eur. J. Org. Chem., 2013, 1228 CrossRef CAS; (b) K. Dahl, M. Schou, O. Rahman and C. Halldin, Eur. J. Org. Chem., 2014, 307 CrossRef CAS; (c) T. Andersen, S. Friis, H. Audrain, P. Nordeman, G. Antoni and T. Skrydstrup, J. Am. Chem. Soc., 2015, 137, 1548 CrossRef CAS PubMed.
- (a) M. Takashima-Hirano, H. Ishii and M. Suzuki, ACS Med. Chem. Lett., 2012, 3, 804 CrossRef CAS PubMed; (b) H. Ishii, K. Minegishi, K. Nagatsu and M. R. Zhang, Tetrahedron, 2015, 10, 1588 CrossRef.
- P. W. Miller, N. Long, A. J. deMello, R. Vilar, H. Audrain, D. Bender, J. Passchier and A. Gee, Angew. Chem., Int. Ed., 2007, 46, 2875 CrossRef CAS PubMed.
- (a) K. Elvira, X. C. Solvas, R. Wootton and A. J. deMello, Nat. Chem., 2013, 5, 905 CrossRef CAS PubMed; (b) G. Whitesides, Nature, 2006, 442, 368 CrossRef CAS PubMed; (c) B. Mason, K. Price, J. Steinbacher, A. Bogdan and D. T. McQuade, Chem. Rev., 2007, 107, 2300 CrossRef CAS PubMed.
- (a) T. Fukuyama, T. Totoki and I. Ryu, Green Chem., 2014, 16, 2042 RSC; (b) A. Günther and K. F. Jensen, Lab Chip, 2006, 6, 1487 RSC; (c) V. van Steijn, M. Kreutzer and C. Kleijn, Chem. Eng. Sci., 2007, 62, 7505 CrossRef CAS; (d) A. Günther, S. Khan, M. Thalmann, F. Trachsel and K. F. Jensen, Lab Chip, 2004, 4, 278 RSC; (e) X. Gong, P. W. Miller, A. Gee, N. Long, A. J. deMello and R. Vilar, Chem.–Eur. J., 2012, 18, 2768 CrossRef CAS PubMed.
- (a) G. Pascali, P. Watts and P. Salvadori, Nucl. Med. Biol., 2013, 6, 776 CrossRef PubMed; (b) A. Elizarov, Lab Chip, 2009, 9, 1326 RSC; (c) J. H. Chun, S. Lu, Y. S. Lee and V. W. Pike, J. Org. Chem., 2010, 75, 3332 CrossRef CAS PubMed; (d) G. Pascali, L. Matesic, T. Collier, N. Wyatt, B. Fraser, T. Pham, P. Salvadori and I. Greguric, Nat. Protoc., 2014, 9, 2017 CrossRef CAS PubMed; (e) S. Liang, T. Collier, B. Rotstein, R. Lewis, M. Steck and N. Vasdev, Chem. Commun., 2013, 49, 8755 RSC; (f) J. M. Gillies, C. Prenant, G. N. Chimon, G. J. Smethurst, W. Perrie, I. Hamblett, B. Dekker and J. Zweit, Appl. Radiat. Isot., 2006, 64, 325 CrossRef CAS PubMed; (g) K. Dahl, M. Schou and C. Halldin, J. Labelled Compd. Radiopharm., 2012, 55, 455 CrossRef CAS; (h) A. Labedev, R. Miraghaie, K. Kotta, C. E. Bell, J. Zhang, M. S. Buchsbaum, H. C. Kolb and A. Elizarov, Lab Chip, 2013, 13, 136 RSC; (i) S. Liang, D. Yokell, M. Normandin, P. Price, R. Jackson, T. Shoup, T. Brandy, G. El Fakhri, T. Collier and N. Vasdev, Mol. Imaging, 2014, 13, 1 CAS; (j) M. Zheng, T. Collier, F. Bois, O. Kelada, K. Hammond, J. Ropchan, M. Akula, G. Kabalka and Y. Huang, Nucl. Med. Biol., 2015, 42, 578 CrossRef CAS PubMed.
- S. Lu, P. Watts, F. Chin, J. M. Musachio, E. Briard and V. W. Pike, Lab Chip, 2004, 4, 523 RSC.
- P. W. Miller, H. Audrain, D. Bender, A. J. deMello and A. Gee, Chem.–Eur. J., 2011, 17, 460 CrossRef CAS PubMed.
- S. Kealey, C. Pilsson, T. Collier, N. Long, S. Husbands, L. Martarello and A. Gee, Org. Biomol. Chem., 2011, 9, 3313 CAS.
- N. Erondu, I. Gantz, B. Musser, S. Suryawanshi, M. Mallick, C. Addy, J. Cote, G. Bray, K. Fujioka, H. Bays, P. Hollander, S. M. Sanabria-Bohórquez, W. Eng, B. Långström, R. J. Hargreaves, H. D. Burns, A. Kanatani, T. Fukami, D. J. MacNeil, K. M. Gottesdiener, J. M. Amatruda, K. D. Kaufman and S. B. Heymsfield, Cell Metab., 2006, 4, 260 CrossRef PubMed.
- C. Halldin, L. Farde, T. Högberg, N. Mohell, H. Hall, T. Suhara, P. Karlsson, Y. Nakashima and C. Swahn, J. Nucl. Med., 1995, 36, 1275 CAS.
- E. Ehrin, L. Farde, T. de Paulis, L. Eriksson, T. Greitz, P. Johnström, J. Nillson, G. Sedvall, S. Stone-Elander and S. Ögren, Int. J. Appl. Radiat. Isot., 1985, 36, 269 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra20646d |
| This journal is © The Royal Society of Chemistry 2015 |