
Biogenic nano-particulate iron-sulfide produced through sulfate and Fe(iii)-(hydr)oxide reductions was enhanced by pyruvate as the electron donor†
Abstract
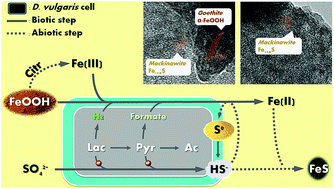
In nature, the formation of iron sulfide solids is mainly attributed to reductions of sulfate and ferric minerals by microorganisms such as Desulfovibrio vulgaris. In order to evaluate the impacts on microbial activity and optimize iron sulfide production for potential application in uranium remediation, we tested two types of electron donors (lactate and pyruvate) with three synthetic Fe(III) (hydr)oxides (goethite, hematite, and 2-line ferrihydrite). We monitored bacterial metabolism comprehensively, and characterized the biogenic solids using transmission electron microscopy equipped with energy-dispersive X-ray spectroscopy (TEM/EDX), X-ray photoelectron spectroscopy (XPS), X-ray diffraction (XRD), Raman spectroscopy, and mass distribution modeling. Despite similar amorphous FeS production when both e− donors were overdosed, D. vulgaris exhibited distinct patterns of metabolism and other solid production with the two electron donors. Once sulfate reduction was complete, further lactate fermentation was inhibited by accumulation of H2, and thus limited FeS production. In contrast, D. vulgaris utilized all pyruvate by diverting electrons from H2 to formate. In addition, the pH decrease due to the proton release during pyruvate utilization facilitated citrate-induced Fe(III) dissolution and consequently enhanced Fe(III) bioavailability. However, higher pH during lactate utilization and excess soluble Fe(II) during pyruvate utilization led to precipitation of Ca3(PO4)2 and Fe3(PO4)2, respectively. Together, these phenomena resulted in a substantial enhancement of Fe(III)-(hydr)oxide reduction and iron sulfide productivity with pyruvate, though the concentrations of calcium and phosphate need to be controlled to avoid precipitation of other minerals.
 Please wait while we load your content...
Please wait while we load your content...

