
Iron-mediated AGET ATRP with crown ether as both ligand and solvent
Abstract
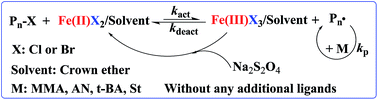
It is well known that crown ethers can selectively complex with metal ions due to their unique structure, and they have been widely used as an ion carrier and in phase-transfer catalysis in organic synthesis. In this work, crown ethers such as 18-crown-6 or 15-crown-5 were introduced into an iron-mediated AGET ATRP (Activators Generated by Electron Transfer for Atom Transfer Radical Polymerization) system as both ligand and solvent for the first time. Herein, FeCl3·6H2O was used as the catalyst, ethyl alpha-bromophenylacetate (EBPA) as the initiator, Na2S2O4 as the reducing agent, and methyl methacrylate (MMA) as the model monomer, and then the method was extended to styrene (St), acrylonitrile (AN) and tert-butyl acrylate (t-BA), without any additional ligands. The effect of various factors, such as the type of reducing agents, the amounts of 18-crown-6, and polymerization temperatures (60–90 °C) on the polymerization were investigated. Furthermore, the polymerization kinetics revealed the typical “living” features of this polymerization system. For example, the molecular weights of PMMA increased linearly with the monomer conversion while maintaining a relatively low molecular weight distribution (Mw/Mn < 1.31). As well as this, the “living” feature of this polymerization system was further confirmed by chain-end analysis (1H NMR, MALDI-TOF) and chain extension experiments.
 Please wait while we load your content...
Please wait while we load your content...

