DOI:
10.1039/C5RA20363E
(Paper)
RSC Adv., 2015,
5, 98033-98040
Convergent synthesis of the hexasaccharide related to the repeating unit of the O-antigen from E. coli O120†
Received
1st October 2015
, Accepted 9th November 2015
First published on 11th November 2015
Abstract
Chemical synthesis of the hexasaccharide, α-L-Rhap-(1→4)-β-D-GlcAp-(1→2)-α-L-Rhap-(1→2)-α-L-Rhap-(1→2)-α-D-Galp-(1→3)-β-D-GalNAcp, is reported by following a [2 + 4] convergent strategy. A disaccharide α-D-Galp-(1→3)-β-D-GalNAcp and a tetrasaccharide α-L-Rhap-(1→4)-β-D-GlcAp-(1→2)-α-L-Rhap-(1→2)-α-L-Rhap were synthesized from commercially available monosaccharides through rational protecting group manipulations and stereoselective glycosylation involving activation of thioglycoside using N-iodosuccinimide and H2SO4-silica. Final glycosylation between the tetrasaccharide donor and the disaccharide acceptor was achieved by the activation of trichloroacetimidate using H2SO4-silica alone. A late stage TEMPO-mediated oxidation installed the required uronic acid moiety. Finally, global deprotection furnished the target molecule. Successful chemical synthesis of the repeating unit of E. coli O120 will help to design suitable vaccine candidates against this deadly pathogen belonging to the STEC family.
Introduction
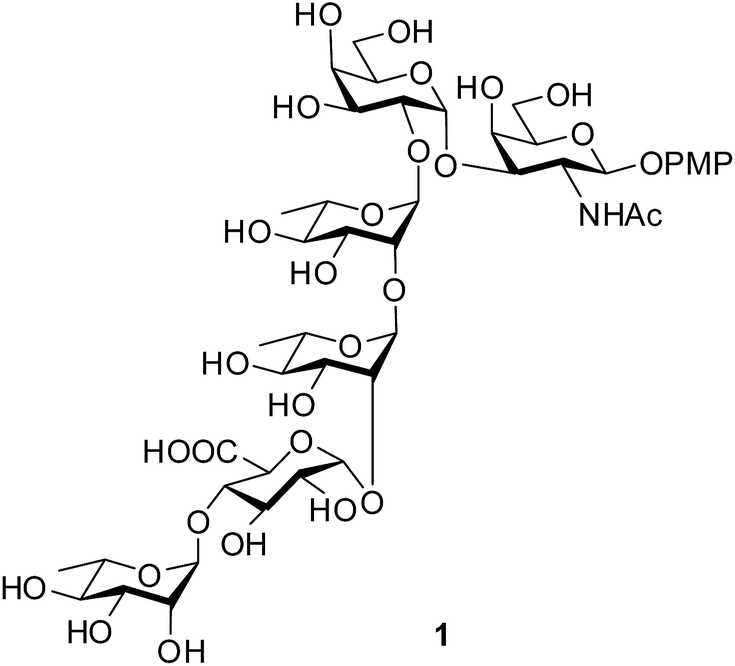
The O-antigen (O-polysaccharide) is an important part of the lipopolysaccharide present on the surface of the Gram-negative bacteria. The O-antigen consists of many repeats of an oligosaccharide called O-units and this is one of the most variable constituents of the cell. The commensal and pathogenic clones of E. coli are normally identified by the combination of their somatic (O), flagellar (H) and capsular (K) antigens.1 A class of E. coli, called as Shiga toxin producing E. coli (STEC), is the causative agent for numerous food-borne disease, haemorrhagic colitis and haemolytic-uremic syndrome in human and cattle.2 Strains of E. coli O120 isolated from swine feces or beef products are identified as STEC.3,4 Recently, Knirel et al. elucidated the structure of the repeating unit of the O-antigen from E. coli O120.5 Herein, we report the concise synthesis of the hexasaccharide repeating unit in the form of its 4-methoxyphenyl glycoside (1, Fig. 1). The concise strategy will give the scope for the preparation of this important oligosaccharide structure in the pure form and in quantity that will pave the path for understanding its role in the pathogenic cycle. Moreover, the selective oxidative removal of the 4-methoxyphenyl group using ceric ammonium nitrate (CAN) followed by formation of corresponding trichloroacetimidate derivative will allow the formation of glycoconjugate with suitable aglycons targeting potential vaccine candidates against this deadly pathogen.
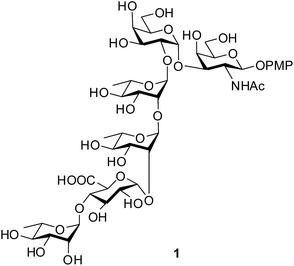 |
| | Fig. 1 Structure of the target hexasaccharide 1. | |
Results and discussion
Retrosynthetic analysis of the hexasaccharide revealed that a convergent [2 + 4] approach will suit best for the synthesis of the linear hexasaccharide. Owing to this, it was first disconnected to a disaccharide, α-D-Galp-(1→3)-β-D-GalNAcp and a tetrasaccharide, α-L-Rhap-(1→4)-α-D-GlcAp-(1→2)-α-L-Rhap-(1→2)-α-L-Rhap. The disaccharide was further disconnected to two monosaccharides that can be derived from commercially available D-GalNH2 and D-Gal. On the otherhand, the tetrasaccharide can be further disconnected to four monosaccharide units, which could be prepared from commercially available D-Glc and three L-Rha moieties through sequential glycosylations. All the monosaccharide derivatives may be obtained from their respective commercially available sugars through rational protecting group manipulations (Fig. 2).
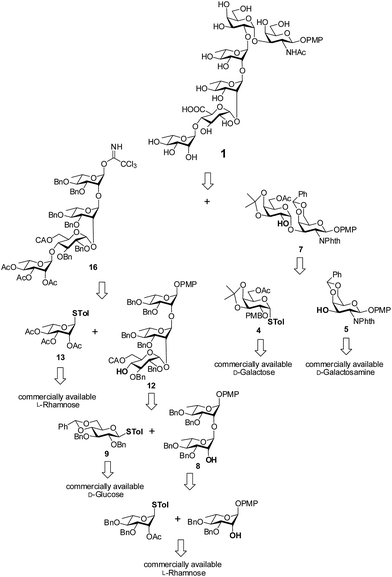 |
| | Fig. 2 Retrosynthetic analysis for the synthesis of the target hexasaccharide 1. | |
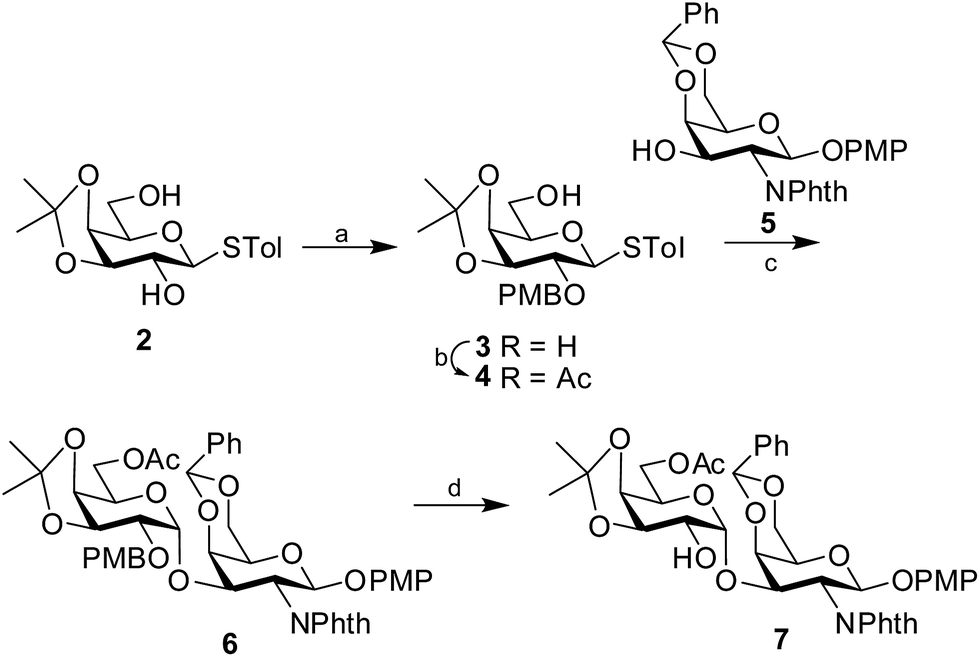
Synthesis of the disaccharide acceptor 7 commenced with the known galactose derivative, 4-tolyl-3,4-O-isopropylidene-1-thio-β-D-galactopyranoside (2).6 Selective alkylation through a phase transfer reaction of compound 2 with 4-methoxybenzyl chloride in presence of TBAB7 incorporated a 4-methoxy benzyl group at the 2-OH position to furnish compound 3. The remaining primary hydroxyl group was further acetylated in the presence of Ac2O in pyridine8 to form the desired galactosyl donor 4 in 94% yield. Glycosylation of the donor 4 with the known galactose acceptor, 4-methoxyphenyl 4,6-O-benzylidene-2-deoxy-2-phthalimido-β-D-galactopyranoside (5)9 through activation of the thioglycoside using NIS in the presence of H2SO4-silica10,11 at −45 °C furnished the disaccharide 6 in 83% isolated yield. Only the desired 1,2-cis disaccharide was formed as evident by the 1H signal at δ 5.0 (d, 1H, J1′,2′ 3.5 Hz) and the 13C signal at δ 95.4 assigned to the newly formed linkage. The exclusive formation of the 1,2-cis linked disaccharide may be explained by the choice of the donor 4 having non-participating 4-methoxybenzyl group at 2-O-position and a α-directive acetyl group at 6-O-position. The 3,4-O-isopropylidene group further provides the required rigidity for α-glycosylation. Finally, oxidative cleavage of the 4-methoxybenzyl group using DDQ12 gave the desired disaccharide acceptor 7 in 81% yield (Scheme 1).
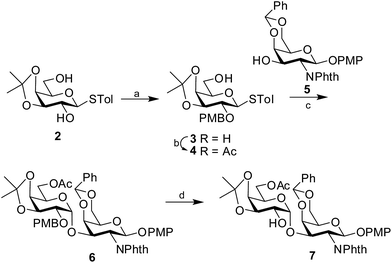 |
| | Scheme 1 Synthesis of the disaccharide acceptor 7. Reagents and conditions: (a) 4-methoxybenzylchloride, TBAB, 87%; (b) Ac2O, Py, 94%; (c) NIS, H2SO4-silica, −45 °C, 83%; (d) DDQ, 81%. | |
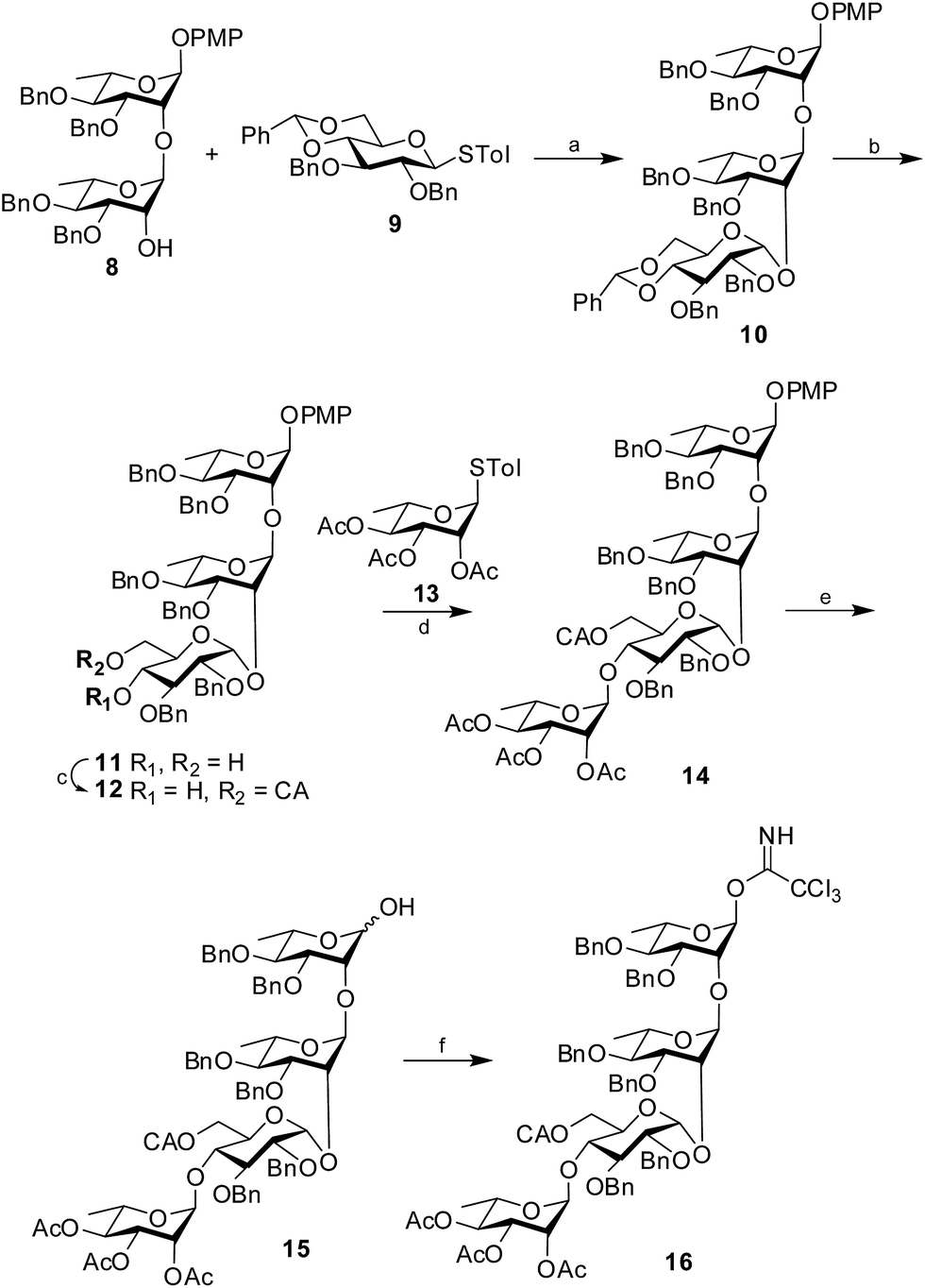
Synthesis of the tetrasaccharide donor 16 commenced with the known disaccharide acceptor 8 (ref. 13) prepared from suitably protected L-rhamnose derivatives following the reported literature procedure. The structure of the disaccharide acceptor was satisfactorily characterized by NMR and mass spectrometry and matched satisfactorily with the data available in the literature. The disaccharide acceptor 8 was then coupled with a known glucose donor, 4,6-O-benzylidene-2,3-di-O-benzyl-β-D-glucopyranoside (9)14 using NIS in the presence of H2SO4-silica at −55 °C to afford the protected trisaccharide 10 in 88% yield. The newly formed 1,2-cis glycosidic linkage was confirmed by the 1H signal at δ 4.83 (d, 1H, J1′′,2′′ 4.5 Hz) and the 13C signal at δ 96.8. The presence of non-participating 2-O-benzyl group and the structural rigidity imposed by 4,6-O-benzylidene group in donor 9 may be attributed for the exclusive formation of the cis-glycoside. Further, the benzylidene ring of the trisaccharide 10 was hydrolyzed using 80% AcOH at 80 °C (ref. 15) to form the diol 11 in 91% yield. Selective chloroacetylation of the primary hydroxyl group was achieved using chloroacetic anhydride and Et3N16 at −5 °C to form the desired trisaccharide acceptor 12 in 89% yield. The trisaccharide acceptor was finally coupled with a known rhamnose donor, 4-tolyl 2,3,4-tri-O-acetyl-1-thio-α-L-rhamnopyranoside (13)17 using NIS in the presence of H2SO4-silica at 0 °C to furnish the protected tetrasaccharide derivative 14 in 85% isolated yield. Only the desired 1,2-trans glycoside was formed as evident by the 1H signal at δ 4.94 and the 13C signal at δ 97.6 assigned to the newly formed linkage. CAN-mediated18 oxidative cleavage of the 4-methoxyphenyl group resulted the corresponding hemiacetal 15 in 78% yield, which was subsequently treated with trichloroacetonitrile in the presence of DBU19 to furnish the tetrasaccharide trichloroacetimidate 16 in 84% yield. Considering the reactivity of the trichloroacetimidate derivative, it was directly used for glycosylation without further characterization (Scheme 2).
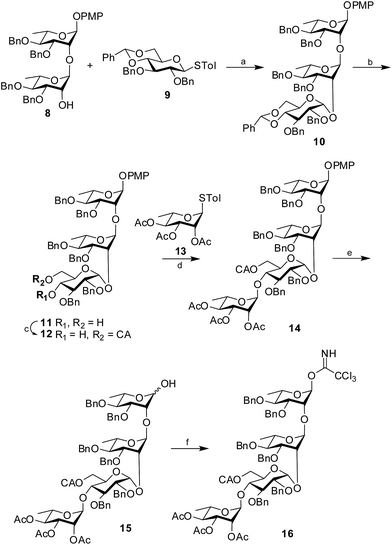 |
| | Scheme 2 Synthesis of the tetrasaccharide donor 16. Reagents and conditions: (a) NIS, H2SO4-silica, −55 °C, 88%; (b) 80% AcOH, 80 °C, 91%; (c) chloroacetic anhydride, Et3N, 89%; (d) NIS, H2SO4-silica, 0 °C, 85%; (e) CAN, CH2Cl2–H2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 78%; (f) CCl3CN, DBU, 84%. :1), 78%; (f) CCl3CN, DBU, 84%. | |
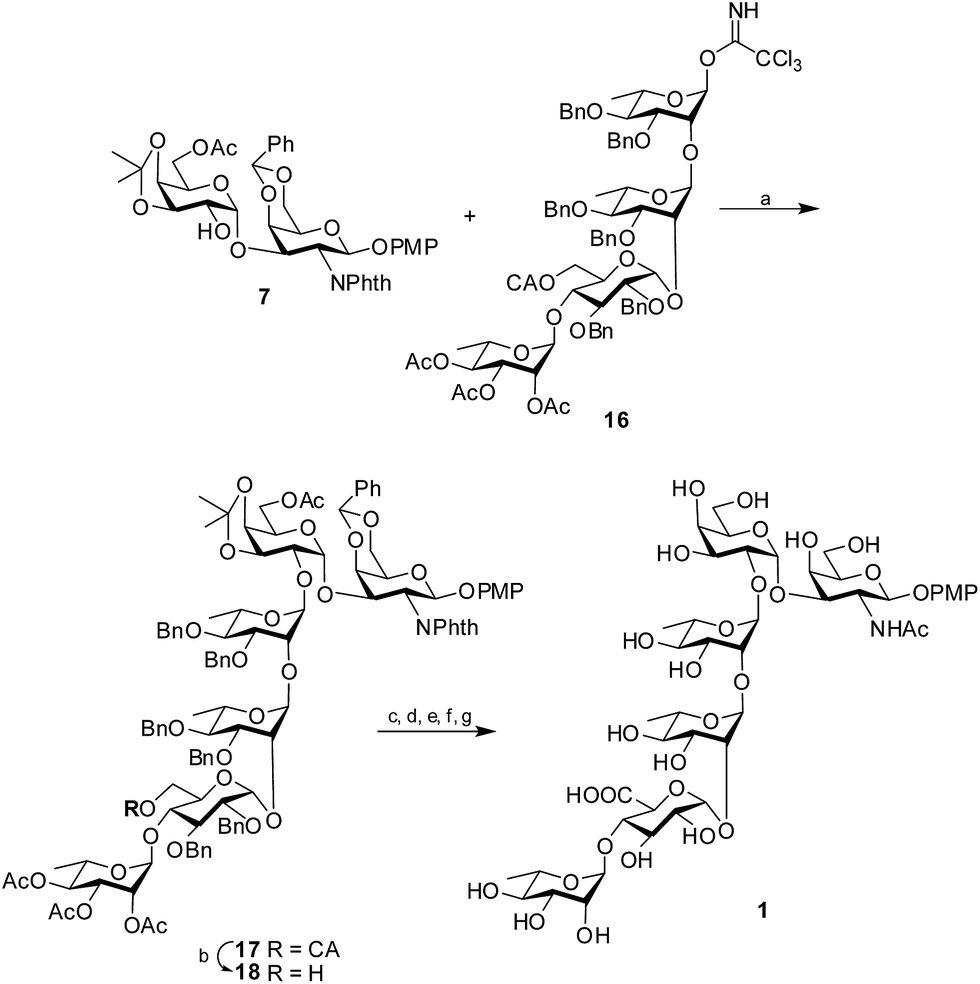
Glycosylation of the disaccharide acceptor 7 with the tetrasaccharide donor 16 through the activation of the trichloroacetimidate by H2SO4-silica20 afforded the protected hexasaccharide 17 in 81% isolated yield. The desired 1,2-trans glycoside was formed solely, as evident by the 1H signal at δ 5.05 and the 13C signal at δ 100.6 assigned to the newly formed linkage. Once the protected hexasaccharide was obtained, the chloroacetate group was selectively deprotected using thiourea and 2,4,6-collidine21 to form the hexasaccharide 18. The primary OH group thus obtained was oxidized to the corresponding uronic acid using TEMPO in the presence of iodosobenzene diacetate.22 It is worth noting that the 4-methoxyphenyl group at the reducing end of the protected hexasaccharide remained unaffected by the TEMPO mediated oxidation. Next, the benzylidene and isopropylidene rings were hydrolyzed by 80% AcOH at 80 °C. Further, the phthalimido functionality was converted to the required acetamido moiety by the treatment of ethylene diamine in presence of n-butanol23 followed by acetylation using Ac2O and pyridine. Subsequently, hydrogenolysis using 10% Pd–C cartridge in a continuous flow hydrogenation assembly followed by Zemplén de-O-acetylation using NaOMe in MeOH24 furnished the target hexasaccharide 1 in 54% overall yield (Scheme 3).
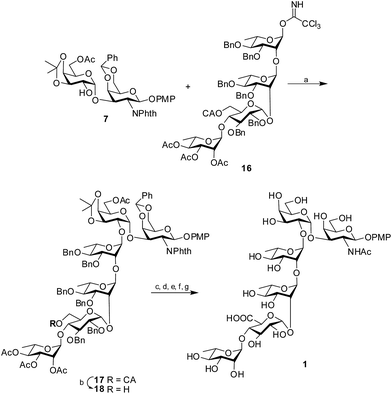 |
| | Scheme 3 Synthesis of the target hexasaccharide 1. Reagents and conditions: (a) H2SO4-silica, 81%; (b) thiourea, 89%; (c) TEMPO, IBDA; (d) 80% AcOH, 80 °C; (e) ethylene diamine, n-butanol; (f) H2, Pd–C; (g) NaOMe, MeOH, 54% overall yield. | |
Conclusions
In conclusion, we have developed a concise strategy for the synthesis of the hexasaccharide repeating unit of the O-antigen from E. coli O120 in the form of its 4-methoxyphenyl glycoside (1). The synthesis of the target compound is achieved through protecting group manipulations on commercially available monosaccharides and stereo-selective glycosylations. The vital glycosylations were achieved either by the activation of thioglycosides using NIS in the presence of H2SO4-silica or by the activation of trichloroacetimidate using H2SO4-silica alone. H2SO4-silica was found to be an efficient alternative to the toxic and hygroscopic acid promoters such as TfOH or TMSOTf. The presence of the 4-methoxyphenyl glycoside would allow selective oxidative deprotection using CAN and further trichloroacetimidate formation to the corresponding hemiacetal. This may open up the path for further glycoconjugate formation with suitable aglycon.
Experimental section
General methods
All solvents and reagents were dried prior to use according to standardized methods.25 The commercially purchased reagents were used without any further purification unless mentioned otherwise. Dichloromethane was dried and distilled over P2O5 to make it anhydrous and moisture-free. All reactions were monitored by Thin Layer Chromatography (TLC) on Silica-Gel 60-F254 with detection by fluorescence followed by charring after immersion in 10% ethanolic solution of H2SO4. Flash chromatography was performed with silica gel 230–400 mesh. Optical rotations were measured on sodium-line at ambient temperature. 1H and 13C NMR were recorded on Bruker 500 MHz spectrometer. 1H NMR values were denoted as H for the reducing end galactosamine unit, H′ for the galactose unit, H′′ for the rhamnose unit connected to the 2-O-position of the galactose unit, H′′′ for the fourth rhamnose unit from the reducing end, H′′′′ for the glucoronic acid moiety linked to the 2-O-position of the rhamnose unit and H′′′′′ for the rhamnose unit present in the non-reducing end.
Preparation of H2SO4-silica
To slurry of silica gel (10 g, 230–400 mesh) in dry diethyl ether (50 mL) was added commercially available concentrated H2SO4-silica (1 mL), and the slurry was shaken for 5 min. The solvent was evaporated under reduced pressure, resulting in free flowing H2SO4-silica, which was dried at 110 °C for 3 h and then used for the reactions.
4-Tolyl 2-O-(4-methoxybenzyl)-3,4-O-isopropylidene-1-thio-β-D-galactopyranoside (3)
Compound 2, 4-tolyl 3,4-O-isopropylidene-1-thio-β-D-galactopyranoside (2 g, 6.1 mmol) was dissolved in CH2Cl2 (40 mL). To it, 10% NaOH solution (5 mL) followed by TBAB (2.2 g, 6.7 mmol) and 4-methoxybenzyl chloride (1.2 mL, 8.6 mmol) added. Reaction continued at room temperature for overnight. The organic layer was washed with water (2 × 50 mL). The organic layer was collected, dried over Na2SO4, evaporated in vacuo and finally purified by flash chromatography using n-hexane–EtOAc (2:1) to afford the pure product 3 (2.4 g, 87%). [α]25D +106 (c 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.45, 7.13 (2d, 4H, J 8.0 Hz, CH2C6H4OCH3), 7.38, 6.91 (2d, 4H, J 9.0 Hz, SC6H4CH3), 4.78, 4.65 (2d, 2H, J 11.0 Hz, CH2C6H4OCH3), 4.59 (d, 1H, J1,2 9.5 Hz, H-1), 4.28 (t, 1H, J2,3, J3,4 6.0 Hz, H-3), 4.19 (dd, 1H, J3,4 6.0 Hz, J4,5 2.0 Hz, H-4), 3.95 (q, 1H, J5,6a 8.5 Hz, J6a,6b 15.0 Hz, H-6a), 3.83 (s, 3H, CH2C6H4OCH3), 3.79 (m, 2H, H-5, H-6b), 3.53 (dd, 1H, J1,2 9.5 Hz, J2,3 6.0 Hz, H-2), 2.35 (s, 3H, SC6H4CH3), 1.44, 1.37 (2s, 6H, 2 × isopropylidene-CH3). 13C NMR (CDCl3, 125 MHz) δ: 159.3, 137.7, 132.5(2), 129.9, 129.8(2), 129.6(2), 129.5, 113.6(2) (ArC), 110.2 [C(CH3)2], 86.2 (C-1), 79.8, 77.9, 76.6, 73.8, 73, 62.5, 55.2 (CH2C6H4OCH3), 27.7, 26.3 (2 × isopropylidene-CH3), 21 (SC6H4CH3). HRMS calcd for C24H30O6SNa (M + Na)+: 469.1661, found: 469.1659.
4-Tolyl 6-O-acetyl-2-O-(4-methoxybenzyl)-3,4-O-isopropylidene-1-thio-β-D-galactopyranoside (4)
Compound 3 (2.4 g, 5.4 mmol) was acetylated using acetic anhydride (10 mL) in presence of pyridine (10 mL). The reaction was completed after 2 h as judged by TLC (n-hexane–EtOAc; 3:2). The solvents were evaporated using toluene as co-solvent. The crude residue was then purified by flash chromatography using n-hexane–EtOAc (4:1) to afford the monosaccharide donor 4 (2.5 g, 94%). [α]25D +121 (c 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.45, 7.09 (2d, 4H, J 8.0 Hz, CH2C6H4OCH3), 7.35, 6.88 (2d, 4H, J 8.5 Hz, SC6H4CH3), 4.75, 4.63 (2d, 2H, J 11.0 Hz, CH2C6H4OCH3), 4.54 (d, 1H, J1,2 9.5 Hz, H-1), 4.32 (m, 2H, H-6), 4.25 (t, 1H, J2,3, J3,4 6.0 Hz, H-3), 4.16 (dd, 1H, J3,4 6.0 Hz, J4,5 2.0 Hz, H-4), 3.89 (m, 1H, H-5), 3.8 (s, 3H, CH2C6H4OCH3), 3.51 (dd, 1H, J1,2 9.5 Hz, J2,3 6.5 Hz, H-2), 2.32 (s, 3H, COCH3), 2.06 (s, 3H, SC6H4CH3), 1.42, 1.34 (2s, 6H, 2 × isopropylidene-CH3). 13C NMR (CDCl3, 125 MHz) δ: 170.7 (COCH3), 159.3, 137.6, 132.6(2), 129.9(4), 129.4(2), 113.7(2) (ArC), 110.3(2) [C(CH3)2], 86.5 (C-1), 79.5, 77.8, 74, 73.5, 73.1, 63.7, 55.2 (CH2C6H4OCH3), 27.7, 26.3 (2 × isopropylidene-CH3), 21.1 (SC6H4CH3), 20.8 (COCH3). HRMS calcd for C26H32O7SNa (M + Na)+: 511.1766, found: 511.1763.
4-Methoxyphenyl 6-O-acetyl-2-O-(4-methoxybenzyl)-3,4-O-isopropylidene-β-D-galactopyranosyl-(1→3)-4,6-O-benzylidene-2-deoxy-2-phthalimido-β-D-galactopyranoside (6)
A mixture of galactose donor 4 (1.3 g, 2.6 mmol), known monosaccharide acceptor, 4-methoxyphenyl 2-phthalimido-4,6-O-benzylidene-2-deoxy-β-D-glucopyranoside 5 (1.0 g, 2 mmol) and MS 4 Å (2.0 g) in dry CH2Cl2 (20 mL) was stirred under nitrogen for 1 h. NIS (750 mg, 3.4 mmol) was added to it and the mixture was cooled to −45 °C followed by addition of H2SO4-silica (50 mg). The mixture was allowed to stir at the same temperature for 1 h when TLC (n-hexane–EtOAc; 1:1) revealed that entire acceptor 5 was consumed. Et3N was added to neutralize the reaction. The reaction mixture was immediately filtered through a pad of Celite. The filtrate was successively washed with Na2S2O3 (2 × 50 mL), NaHCO3 (2 × 50 mL) and brine (50 mL). The organic layer was collected, dried (Na2SO4) and evaporated in vacuo. The crude product thus obtained was purified by flash chromatography using n-hexane–EtOAc (3:2) to give the pure disaccharide 6 (1.3 g, 83%) as a white foam. [α]25D +86 (c 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.85–7.06 (m, 13H, ArH), 6.88, 6.71 (2d, 4H, J 9.0 Hz, C6H4OCH3), 5.73 (d, 1H, J1,2 8.5 Hz, H-1), 5.5 (s, 1H, CHPh), 5.0 (d, 1H, J1′,2′ 3.5 Hz, H-1′), 4.97 (dd, 1H, J1,2 8.5 Hz, J2,3 11.5 Hz, H-2), 4.78 (dd, 1H, J2,3 11.0 Hz, J3,4 3.5 Hz, H-3), 4.52 (bs, 2H, CH2C6H4OCH3), 4.41 (d, 1H, J3,4 3.5 Hz, H-4), 4.37 (dd, 1H, J5,6a′ 1.5 Hz, J6a′,6b′ 12.0 Hz, H-6a′), 4.18 (t, J2′,3′, J3′,4′ 6.0 Hz, H-3′), 4.06 (dd, 1H, J5,6b′ 1.5 Hz, J6a′,6b′ 12.0 Hz, H-6b′), 3.92 (m, 3H, H-4′, H-6), 3.76 (s, 3H, CH2C6H4OCH3), 3.71 (m, 4H, H-5, C6H4OCH3), 3.62 (bs, 1H, H-5′), 3.56 (dd, 1H, J1′,2′ 3.5 Hz, J2′,3′ 6.0 Hz, H-2′), 2.73 (s, 6H, 2 × isopropylidene-CH3), 2.02 (s, 3H, COCH3). 13C NMR (CDCl3, 125 MHz) δ: 170.4 (COCH3), 168.8, 167.5 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of NPhth), 159.1, 155.4, 150.3, 137.7, 134.1, 134.0, 131.6, 131.5, 130.2, 129.5(2), 128.9, 128.1(2), 128(2), 126.4(2), 123.6, 123.2, 119.2(2), 114.3(2), 113.6(2) (ArC), 109.5 [C(CH3)2], 100.9 (CHPh), 98.2 (C-1), 95.4 (C-1′), 74.7, 73.6, 72.6, 72.5, 72.4, 72.2, 69.2, 67.3, 66.8, 62.6, 55.5 (C6H4OCH3), 55.2 (CH2C6H4OCH3), 52.2 (C-2), 27.2, 25.6 (2 × isopropylidene-CH3), 20.8 (COCH3). HRMS calcd for C47H49O15NNa (M + Na)+: 890.3000, found: 890.2997.
O of NPhth), 159.1, 155.4, 150.3, 137.7, 134.1, 134.0, 131.6, 131.5, 130.2, 129.5(2), 128.9, 128.1(2), 128(2), 126.4(2), 123.6, 123.2, 119.2(2), 114.3(2), 113.6(2) (ArC), 109.5 [C(CH3)2], 100.9 (CHPh), 98.2 (C-1), 95.4 (C-1′), 74.7, 73.6, 72.6, 72.5, 72.4, 72.2, 69.2, 67.3, 66.8, 62.6, 55.5 (C6H4OCH3), 55.2 (CH2C6H4OCH3), 52.2 (C-2), 27.2, 25.6 (2 × isopropylidene-CH3), 20.8 (COCH3). HRMS calcd for C47H49O15NNa (M + Na)+: 890.3000, found: 890.2997.
4-Methoxyphenyl 6-O-acetyl-3,4-O-isopropylidene-β-D-galactopyranosyl-(1→3)-4,6-O-benzylidene-2-deoxy-2-phthalimido-β-D-galactopyranoside (7)
Disaccharide 6 (1.3 g, 1.5 mmol) was dissolved in CH2Cl2–H2O (20:5) mixture. DDQ (660 mg, 2.9 mmol) was added to the reaction mixture and it was kept stirring for 2 h, as monitored by TLC (n-hexane–EtOAc; 1:1). The reaction mixture was then successively washed with H2O (2 × 50 mL). The organic layer was collected, dried over Na2SO4, filtered and evaporated in vacuo. The product, thus obtained, was purified using flash chromatography using n-hexane–EtOAc (1:1) as eluent to furnish the disaccharide acceptor 7 (910 mg, 81%) in pure form. [α]25D +108 (c 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.9–7.38 (m, 9H, ArH), 6.9, 6.73 (2d, 4H, J 9.5 Hz, C6H4OCH3), 5.83 (d, 1H, J1,2 8.0 Hz, H-1), 5.62 (s, 1H, CHPh), 5.04 (d, 1H, J1′,2′ 4.0 Hz, H-1′), 4.86 (dd, 1H, J1,2 8.0 Hz, J2,3 11.0 Hz, H-2), 4.79 (dd, 1H, J2,3 11.0 Hz, J3,4 4.0 Hz, H-3), 4.44 (d, 1H, J3,4 3.5 Hz, H-4), 4.41 (dd, 1H, J5,6a′ 1.0 Hz, J6a′,6b′ 12.5 Hz, H-6a′), 4.13 (dd, 1H, J5,6b′ 1.5 Hz, J6a′,6b′ 12.0 Hz, H-6b′), 4.07 (t, J2′,3′, J3′,4′ 6.0 Hz, H-3′), 3.84 (m, 2H, H-6), 3.82 (dd, 1H, J3′,4′ 6.0 Hz, J4′,5′ 2.5 Hz, H-4′), 3.71 (m, 2H, H-2, C6H4OCH3), 3.67 (m, 1H, H-5), 3.65 (m, 1H, H-5′), 2.0 (s, 3H, COCH3), 1.41, 1.22 (2s, 6H, 2 × isopropylidene-CH3). 13C NMR (CDCl3, 125 MHz) δ: 170.2 (COCH3), 168.4, 167.7 (CO of NPhth), 155.6, 150.7, 137.2, 134.3, 131.4, 131.3, 129.2, 128.4(2), 128.3, 126.3(2), 123.7, 123.4, 119.3(3), 114.4(2) (ArC), 109.7 [C–(CH3)2], 101.3 (CHPh), 97.9 (C-1), 94.5 (C-1′), 75.4, 72.3, 71.8, 71.6, 69.2, 68.6, 66.9, 66.6, 62.7, 55.5 (C6H4OCH3), 52.1 (C-2), 27.4, 25.7 (2 × isopropylidene-CH3), 20.8 (COCH3). HRMS calcd for C39H41O14NNa (M + Na)+: 770.2425, found: 770.2422.
4-Methoxyphenyl 4,6-O-benzylidene-2,3-di-O-benzyl-β-D-glucopyranosyl (1→2)-3,4-di-O-benzyl-α-L-rhamnopyranosyl (1→2)-3,4-di-O-benzyl-α-L-rhamnopyranoside (10)
To a mixture of disaccharide acceptor 8 (2.0 g, 2.6 mmol), known donor 4-tolyl 4,6-O-benzylidene-2,3-di-O-benzyl-1-thio-β-D-glucopyranoside (9) (1.9 g, 3.4 mmol) in dry CH2Cl2 (30 mL) and 4 Å powdered molecular sieves (3.0 g) was added and the mixture was stirred under nitrogen atmosphere for 1 h. NIS (1.0 g, 4.4 mmol) was added to the reaction mixture and it was cooled to −55 °C. H2SO4-silica (50 mg) was added to the reaction mixture and it was stirred for 25 min until TLC (hexane–EtOAc; 2:1) indicated complete consumption of acceptor 8. The reaction mixture was diluted with CH2Cl2 (30 mL) and filtered through a pad of Celite. The filtrate was then successively washed with aq. Na2S2O3 (2 × 50 mL), saturated aq. NaHCO3 (2 × 50 mL) and brine (50 mL). The organic layer was separated, dried (Na2SO4) and evaporated in vacuo. The crude trisaccharide thus obtained, was purified by flash chromatography using n-hexane–EtOAc (3:1) as eluent to afford pure trisaccharide 10 (2.7 g, 88%). [α]25D +78 (c 0.9, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.57–7.28 (m, 35H, ArH), 6.99, 6.86 (2d, 4H, J 9.0 Hz, C6H4OCH3), 5.61 (s, 1H, CHPh), 5.38 (d, 1H, J1,2 1.5 Hz, H-1), 5.11 (d, 1H, J1′,2′ 1.5 Hz, H-1′), 4.96 (m, 3H, CH2Ph), 4.84 (m, 2H, CH2Ph), 4.83 (d, 1H, J1′′,2′′ 4.5 Hz, H-1′′), 4.77, 4.64 (2d, 2H, J 12.0 Hz, CH2Ph), 4.76, 4.59, 4.44 (3d, 3H, J 11.5 Hz, CH2Ph), 4.7, 4.68 (2d, 2H, J 11.0 Hz, CH2Ph), 4.32 (m, 1H, H-5′′), 4.2 (t, 1H, J1,2, J2,3 2.5 Hz, H-2), 4.18 (t, 1H, J1′,2′, J2′,3′ 2.5 Hz, H-2′), 4.13 (m, 2H, H-3′′, H-4′′), 4.06 (dd, 1H, J2′,3′ 3.0 Hz, J3′,4′ 9.5 Hz, H-3′), 3.99 (dd, 1H, J2,3 3.0 Hz, J3,4 9.5 Hz, H-3), 3.85 (m, 2H, H-5, H-5′), 3.81 (s, 3H, C6H4OCH3), 3.66 (m, 3H, H-4, H-6′′), 3.49 (m, 2H, H-2′′, H-4′), 1.35 (d, 3H, J5,6 6.0 Hz, C–CH3), 1.3 (d, 3H, J5′,6′ 6.5 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 154.8, 150.2, 138.9, 138.6, 138.4, 138.3, 138.2, 137.6, 128.8, 128.4(3), 128.3(3), 128.2(3), 128.1(3), 128.0(3), 127.9(3), 127.8(3), 127.7(3), 127.6(2), 127.5(3), 127.4, 127.3, 127.2(2), 126.1(2), 117.4(2), 114.6(2) (ArC), 101.2 (CHPh), 98.8 (C-1′), 97.7 (C-1), 96.8 (C-1′′), 82.4, 80.4, 80.1, 79.6, 79.3, 78.1, 78, 75.3, 75, 74.3, 74.2, 72.3, 72.2(2), 69.1, 68.9, 68.5, 62.5, 55.6 (C6H4OCH3), 18.0(2) (2 × C–CH3). HRMS calcd for C74H78O15Na (M + Na)+: 1229.5238, found: 1229.5235.
4-Methoxyphenyl 2,3-di-O-benzyl-β-D-glucopyranosyl (1→2)-3,4-di-O-benzyl-α-L-rhamnopyranosyl (1→2)-3,4-di-O-benzyl-α-L-rhamnopyranoside (11)
The trisaccharide 10 (2.7 g, 2.2 mmol) was treated with 80% AcOH (20 mL) at 80 °C for 2 h resulting in cleavage of benzylidene ring. The solvents were then evaporated using toluene as co-solvent and the reaction mixture was dried in vacuum. The product was then purified by flash chromatography using n-hexane–EtOAc (1:1) as eluent to furnish pure product 11 (2.3 g, 91%). [α]25D +131 (c 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.47–7.22 (m, 30H, ArH), 6.99, 6.86 (2d, 4H, J 9.0 Hz, C6H4OCH3), 5.39 (d, 1H, J1,2 1.5 Hz, H-1), 5.16 (s, 1H, H-1′), 4.97 (m, 3H, CH2Ph), 4.87 (d, 1H, J1′′,2′′ 3.5 Hz, H-1′′), 4.81, 4.76 (2d, 2H, J 12.0 Hz, CH2Ph), 4.77 (d, 2H, J 11.5 Hz, CH2Ph), 4.71, 4.69 (2d, 2H, J 11.0 Hz, CH2Ph), 4.66 (d, 1H, J 11.5 Hz, CH2Ph), 4.45 (bs, 2H, CH2Ph), 4.21 (t, 1H, J1,2, J2,3 2.5 Hz, H-2), 4.2 (t, 1H, J1′,2′, J2′,3′ 2.5 Hz, H-2′), 4.07 (dd, 1H, J2,3 3.0 Hz, J3,4 9.0 Hz, H-3), 4.04 (m, 1H, H-5′′), 4.0 (dd, 1H, J2′,3′ 3.0 Hz, J3′,4′ 9.5 Hz, H-3′), 3.9 (d, 1H, J2′′,3′′ 9.0 Hz, H-3′′), 3.85 (m, 2H, H-5, H-5′), 3.8 (s, 3H, C6H4OCH3), 3.65 (m, 2H, H-4′, H-6a′′), 3.56 (m, 2H, H-4′′, H-6b′′), 3.51 (t, 1H, J3,4, J4,5 9.5 Hz, H-4), 3.44 (dd, 1H, J1′′,2′′ 3.5 Hz, J2′′,3′′ 9.5 Hz, H-2′′), 2.56, 2 (2bs, 2H, 2 × CH2OH), 1.38 (d, 3H, J5,6 6.0 Hz, C–CH3), 1.31 (d, 3H, J5′,6′ 6.0 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 154.8, 150.1, 138.7, 138.4, 138.3, 138.1, 128.5(3), 128.4(3), 128.3(3), 128.2(3), 128.1(3), 128.0(3), 127.8(3), 127.7(3), 127.6(3), 127.5(2), 127.4(3), 117.3(2), 114.6(2) (ArC), 98.4 (C-1′), 97.6 (C-1), 95.0 (C-1′′), 80.7, 80.4, 80.3, 79.7, 79.6, 78.1, 75.3, 75, 74, 73.4, 72.4, 72.1, 71.7, 71, 70.5, 68.9, 68.5, 62.4, 60.3, 55.6 (C6H4OCH3). 18.1, 18.0 (2 × C–CH3). HRMS calcd for C67H74O15Na (M + Na)+: 1141.4925, found: 1141.4923.
4-Methoxyphenyl 2,3-di-O-benzyl-6-O-chloroacetyl-β-D-glucopyranosyl (1→2)-3,4-di-O-benzyl-α-L-rhamnopyranosyl (1→2)-3,4-di-O-benzyl-α-L-rhamnopyranoside (12)
Trisaccharide 11 (2.3 g, 2.1 mmol) was dissolved in dry CH3CN (20 mL) and Et3N (0.3 mL, 2.1 mmol) and stirred at −5 °C for 10 minutes. Chloroacetic anhydride (390 mg, 2.3 mmol) was added and the solution was stirred at −5 °C for 1 hour when TLC (n-hexane–EtOAc; 3:2) revealed complete conversion of the starting material. The solvents were evaporated and coevaporated with toluene for complete removal of pyridine. The crude residue thus obtained was purified by flash chromatography using n-hexane–EtOAc (3:1) as eluent to afford the desired trisaccharide acceptor 12 (2.3 g, 91%). [α]25D +84 (c 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.46–7.30 (m, 30H, ArH), 7.02, 6.89 (2d, 4H, J 9.0 Hz, C6H4OCH3), 5.43 (d, 1H, J1,2 1.5 Hz, H-1), 5.14 (d, 1H, J1′,2′ 1.0 Hz, H-1′), 5.01 (m, 3H, CH2Ph), 4.92 (d, 1H, J1′′,2′′ 3.0 Hz, H-1′′), 4.86–4.68 (m, 7H, CH2Ph), 4.43 (s, 2H, CH2Ph), 4.29 (m, 3H, H-2′, H-5′′, H-6a′′), 4.24 (t, 1H, J1,2, J2,3 2.0 Hz, H-2), 4.15 (m, 1H, H-6b′′), 4.11 (dd, 1H, J2,3 2.5 Hz, J3,4 9.0 Hz, H-3), 4.04 (m, 3H, H-3′, COCH2Cl), 3.89 (m, 3H, H-3′′, H-5, H-5′), 3.82 (s, 3H, C6H4OCH3), 3.69 (t, 1H, J3′,4′, J4′,5′ 9.5 Hz, H-4′), 3.55 (m, 2H, H-4, H-4′′), 3.47 (dd, 1H, J1′′,2′′ 3.5 Hz, J3′′,4′′ 9.5 Hz, H-2′′), 1.42 (d, 3H, J5,6 6.5 Hz, C–CH3), 1.36 (d, 3H, J5′,6′ 6.5 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 167.3 (COCH2Cl), 154.7, 150.1, 138.5, 138.3(3), 138.2(2), 138.1, 128.4(5), 128.3(5), 128.2(2), 128.1(2), 128(2), 127.8(2), 127.7(4), 127.6, 127.5(2), 127.3(2), 127.1(2), 117.3(2), 114.5(2) (ArC), 98.4 (C-1′), 97.6 (C-1), 95 (C-1′′), 80.5, 80.3(2), 79.6, 79.5, 78, 75.4, 75.3, 75.1, 74, 73.5, 72.4, 72.3, 71.7, 69.5, 69, 68.8, 68.5, 64.5, 55.5 (C6H4OCH3), 40.6 (COCH2Cl), 18.0(2) (2 × C–CH3). HRMS calcd for C69H75O16ClNa (M + Na)+: 1217.4641, found: 1217.4638.
4-Methoxyphenyl 2,3,4-tri-O-acetyl-α-L-rhamnopyranosyl-(1→4)-2,3-di-O-benzyl-6-O-chloroacetyl-β-D-glucopyranosyl-(1→2)-3,4-di-O-benzyl-α-L-rhamnopyranosyl-(1→2)-3,4-di-O-benzyl-α-L-rhamnopyranoside (14)
A suspension of trisaccharide acceptor 12 (2.3 g, 1.95 mmol), the known rhamnose donor, 4-tolyl 2,3,4-tri-O-acetyl-1-thio-α-L-rhamnopyranoside 13 (1.0 g, 2.5 mmol) and MS 4 Å (2 g) in dry CH2Cl2 (20 mL) was stirred under nitrogen for 30 minutes. After adding NIS (740 mg, 3.3 mmol), the mixture was cooled to 0 °C. After 10 minutes, H2SO4-silica (50 mg) was added and the mixture was stirred at 0 °C for 50 minutes when TLC (n-hexane–EtOAc; 2:1) revealed complete consumption of the donor. The reaction mixture was then immediately filtered through a pad of Celite and washed with CH2Cl2. The combined filtrate obtained was washed successively with Na2S2O3 (2 × 50 mL), NaHCO3 (2 × 50 mL) and finally with brine (50 mL). The organic layer was separated, dried (Na2SO4), filtered and evaporated in vacuo. The residue was then purified by flash chromatography using n-hexane–EtOAc (3:1) as eluent to give the disaccharide 14 (2.3 g, 85%) as white foam. [α]25D +148 (c 0.9, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.5–7.23 (m, 30H, ArH), 7.02, 6.88 (2d, 4H, J 9.0 Hz, C6H4OCH3), 5.42 (d, 1H, J1,2 1.5 Hz, H-1), 5.3 (dd, 1H, J2′′′,3′′′ 3.5 Hz, J3′′′,4′′′ 10.0 Hz, H-3′′′), 5.16 (d, 1H, J1′,2′ 1.5 Hz, H-1′), 5.06 (m, 5H, H-4′′′, CH2Ph), 4.94 (m, 2H, H-1′′′, CH2Ph), 4.8 (m, 5H, H-1′′, H-2′′′, CH2Ph), 4.67 (d, 1H, J 12.0 Hz, CH2Ph), 4.38 (dd, 1H, J5′′,6a′′ 2.0 Hz, J6a′′,6b′′ 12.5 Hz, H-6a′′), 4.31 (m, 3H, CH2Ph), 4.24 (m, 2H, H-2, H-2′), 4.16 (m, 2H, H-5′, H-5′′′), 4.1 (m, 3H, COCH2Cl, H-3′), 4.05 (dd, 1H, J2,3 3.5 Hz, J3,4 9.5 Hz, H-3), 3.94 (t, 1H, J2′′,3′′, J3′′,4′′ 9.0 Hz, H-3′′), 3.88 (m, 2H, H-5, H-5′′), 3.8 (s, 3H, C6H4OCH3), 3.74 (m, 3H, H-4′, H-4′′, H-6b′′), 3.59 (t, 1H, J3,4, J4,5 9.5 Hz, H-4), 3.46 (dd, 1H, J1′′,2′′ 3.5 Hz, J2′′,3′′ 9.0 Hz, H-2′′), 2.16 (s, 3H, COCH3), 2.08 (bs, 6H, 2 × COCH3), 1.43 (d, 3H, J5,6 6.0 Hz, C–CH3), 1.37 (d, 3H, J5′,6′ 6.5 Hz, C–CH3), 0.98 (d, 3H, J5′′′,6′′′ 6.5 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 170, 169.9, 169.7 (3 × COCH3), 166.6 (COCH2Cl), 154.7, 150, 138.4, 138.3(2), 138.2(4), 138.1(2), 138.0(2), 128.4, 128.3(4), 128.2(2), 128.1, 128(4), 127.9(2), 127.8, 127.7(3), 127.6(2), 127.5, 127.4, 127.3, 127.2, 126.9, 117.2(2), 114.5(2) (ArC), 98.1 (C-1′), 97.6(2) (C-1, C-1′′′), 94.1 (C-1′′), 80.4, 80.3, 80.2, 79.6, 79.2, 77.9, 75.3, 75.2, 74.8, 73.7, 73.1, 72.5, 72.2, 71.7, 70.8, 69.6, 68.8, 68.7, 68.5, 67.9, 66.9, 63.6, 60.2, 55.4 (C6H4OCH3), 40.6 (COCH2Cl), 20.7, 50.6, 20.5(3 × COCH3), 17.9(2) (2 × C–CH3), 17.0 (C–CH3). HRMS calcd for C81H91O23ClNa (M + Na)+: 1489.5537, found: 1489.5533.
4-Methoxyphenyl 2,3,4-tri-O-acetyl-α-L-rhamnopyranosyl (1→4)-2,3-di-O-benzyl-6-O-chloroacetyl-β-D-glucopyranosyl (1→2)-3,4-di-O-benzyl-α-L-rhamnopyranosyl (1→2)-3,4-di-O-benzyl-α-L-rhamnopyranosyl (1→2)-6-O-acetyl-3,4-O-isopropylidene-β-D-galactopyranosyl-(1→3)-4,6-O-benzylidene-2-deoxy-2-phthalimido-β-D-galactopyranoside (17)
A mixture of disaccharide acceptor 7 (500 mg, 0.7 mmol), trichloroacetamidate donor 16 (1.3 g, 0.87 mmol) and freshly activated MS 4 Å (1.0 g) in dry CH2Cl2 (10 mL) was stirred under nitrogen for 1 hour at room temperature. After cooling the reaction mixture to 0 °C, H2SO4-silica (50 mg) was added and the mixture was allowed to stir at the same temperature for 30 min when TLC (n-hexane–EtOAc; 1:1) showed complete consumption of acceptor 7. The reaction mixture was neutralized with Et3N and filtered through a pad of Celite. The filtrate was diluted with CH2Cl2 and was successively washed with NaHCO3 (2 × 50 mL) and brine (50 mL). The organic layer was separated, dried (Na2SO4) and evaporated in vacuo. The crude product thus obtained was purified by flash chromatography using n-hexane–EtOAc (1.2:1) to give the pure hexasaccharide 17 (1.0 g, 81%). [α]25D +124 (c 0.8, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.89–7.05 (m, 39H, ArH), 6.82, 6.69 (2d, 4H, J 9.0 Hz, C6H4OCH3), 5.64 (d, 1H, J1,2 8.5 Hz, H-1), 5.6 (s, 1H, CHPh), 5.2 (dd, 1H, J2′′′′′,3′′′′′ 3.5 Hz, J3′′′′′,4′′′′′ 10.0 Hz, H-3′′′′′), 5.05 (m, 2H, H-1′′, H-4′′′′′), 4.92 (m, 9H, H-1′, H-1′′′, H-1′′′′′, H-2, H-2′′′′′, CH2Ph), 4.71 (m, 5H, H-3, CH2Ph), 4.59 (d, 1H, J1′′′′,2′′′′ 3.5 Hz, H-1′′′′), 4.27 (m, 3H, H-4, H-6a′′′′, CH2Ph), 4.11 (m, 8H, H-2′′, H-2′′′, H-3′, H-5′′′′, H-5′′′′′), 4.03 (bs, 2H, COCH2Cl), 3.91 (m, 2H, H-4′, H-5), 3.83 (m, 3H, H-3′′′′, H-5′′, H-6a′), 3.77–3.59 (m, 12H, H-2′, H-3′′, H-3′′′, H-4′′′′, H-5′, H-5′′′, H-6a, H-6b′, H-6b′′′′, C6H4OCH3), 3.49 (bs, 1H, H-6b), 3.44 (m, 2H, H-4′′, H-4′′′), 3.3 (dd, 1H, J1′′′′,2′′′′ 3.5 Hz, J2,3′′′′ 9.0 Hz, H-2′′′′), 2.11, 2.03, 2.02, 1.99 (4s, 12H, 4 × COCH3), 1.39, 1.19 (2s, 6H, 2 × isopropylidene-CH3), 1.37 (d, 3H, J5′′,6′′ 6.5 Hz, C–CH3), 1.35 (d, 3H, J5′′′,6′′′ 6.5 Hz, C–CH3), 0.89 (d, 3H, J5′′′′′,6′′′′′ 5.5 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 170.2, 170.1, 170, 169.8 (4 × COCH3), 168.9 (COCH2Cl), 167.5, 166.7 (2 × CO of NPhth), 155.5, 150.8, 138.5, 138.4, 138.3, 138.2, 138.1, 137.8, 134.4, 134.3, 131.5, 128.8, 128.5(2), 128.4(3), 128.3(3), 128.2(4), 128.1(5), 128(2), 127.9(2), 127.8, 127.7, 127.6, 127.5, 127.4, 127.3(2), 126.3, 126.2(2), 124.4, 124.3, 124, 123.6, 123.5, 123.4, 119.2(2), 114.3(2) (ArC), 109.1 [C–(CH3)2], 100.6 (C-1′′), 100.2 (CHPh), 99.6 (C-1′′′), 98.1 (C-1), 97.9 (C-1′), 97.6 (C-1′′′′′), 93.8 (C-1′′′′), 80.5, 80.4, 80.2, 79.9, 79.3, 78, 76, 75.9, 75.6, 75.3, 75.2, 74.9, 74.8, 73.5, 72.8, 72.7, 72.6, 72.5, 71.6, 71(2), 69.7, 68.9, 68.8, 68.7, 68.3, 67.8, 67.0, 66.8, 66.4, 63.7, 62.2, 55.5 (C6H4OCH3), 52.3 (C-2), 40.8 (COCH2Cl), 28.2, 26.2 (2 × isopropylidene-CH3), 20.9, 20.8(2), 20.7 (4 × COCH3), 18.1, 17.9, 17.1 (3 × C–CH3). HRMS calcd for C113H124O35ClNNa (M + Na)+: 2112.7540, found: 2112.7536.
4-Methoxyphenyl 2,3,4-tri-O-acetyl-α-L-rhamnopyranosyl (1→4)-2,3-di-O-benzyl-β-D-glucopyranosyl (1→2)-3,4-di-O-benzyl-α-L-rhamnopyranosyl (1→2)-3,4-di-O-benzyl-α-L-rhamnopyranosyl (1→2)-6-O-acetyl-3,4-O-isopropylidene-β-D-galactopyranosyl-(1→3)-4,6-O-benzylidene-2-deoxy-2-phthalimido-β-D-galactopyranoside (18)
To a solution of the hexasaccharide 17 (1.0 g, 0.5 mmol) in CH2Cl2–CH3OH (2:3, 20 mL), thiourea (180 mg, 2.4 mmol) and collidine (0.3 mL, 2.4 mmol) were added and it was refluxed at 40 °C for 10 hours when TLC (n-hexane–EtOAc; 1:1) showed complete conversion of the starting material to a slower moving spot. After evaporating the solvents in vacuo, the residue was dissolved in CH2Cl2 (50 mL) and washed with 1 M HCl (50 mL) and H2O (50 mL). The organic layer was collected, dried (Na2SO4), filtered and evaporated. The crude product thus obtained was purified by flash chromatography using n-hexane–EtOAc (1:1) as eluent to afford the pure hexasaccharide acceptor 18 (850 mg, 89%) as colourless foam. [α]25D +101 (c 0.7, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.89–7.06 (m, 39H, ArH), 6.82, 6.68 (2d, 4H, J 9.0 Hz, C6H4OCH3), 5.64 (d, 1H, J1,2 9.0 Hz, H-1), 5.59 (s, 1H, CHPh), 5.25 (dd, 1H, J2′′′′′,3′′′′′ 3.5 Hz, J3′′′′′,4′′′′′ 10.0 Hz, H-3′′′′′), 5.12 (dd, 1H, J1′′′′′,2′′′′′ 1.5 Hz, J2′′′′′,3′′′′′ 3.5 Hz, H-2′′′′′), 5.05 (d, 1H, J1′′,2′′ 1.5 Hz, H-1′′), 4.95 (m, 6H, H-1′′′, H-2, H-4′′′′′, CH2Ph), 4.87 (s, 1H, H-1′′′′′), 4.85 (d, 1H, J1′,2′ 3.5 Hz, H-1′), 4.78 (d, 1H, J 12.0 Hz, CH2Ph), 4.67 (m, 7H, H-1′′′′, H-3, CH2Ph), 4.29 (d, 1H, J3,4 3.0 Hz, H-4), 4.23 (m, 2H, CH2Ph), 4.15 (m, 2H, H-3′, CH2Ph), 4.06 (m, 3H, H-2′′, H-2′′′, H-5′′′′′), 3.91 (m, 2H, H-4′, H-5′′′′), 3.82 (m, 4H, H-3′′, H-3′′′, H-3′′′′, H-6a′), 3.75 (m, 1H, H-5′′′), 3.65 (m, 10H, H-2′, H-4′′′′, H-5, H-5′, H-5′′, H-6′′′′, C6H4OCH3), 3.49 (bs, 1H, H-6a), 3.42 (m, 3H, H-4′′, H-4′′′, H-6b′), 3.35 (dd, 1H, J1′′′′,2′′′′ 3.5 Hz, J2′′′′,3′′′′ 9.0 Hz, H-2′′′′), 3.24 (d, 1H, J6a,6b 12 Hz, H-6b), 2.11, 2.02, 2.00, 1.99 (4s, 12H, 4 × COCH3), 1.39, 1.19 (2s, 6H, 2 × isopropylidene-CH3), 1.34 (m, 6H, 2 × C–CH3), 0.79 (d, 3H, J5′′′′′,6′′′′′ 6.0 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 170.2, 170.1, 170, 169.9 (4 × COCH3), 168.9, 167.5 (2 × CO of NPhth), 157.1, 155.5, 150.8, 147.9, 138.7, 138.5, 138.4, 138.3, 138.2, 138.1, 137.8, 134.4, 134.3, 131.5, 128.8, 128.4(3), 128.3(2), 128.2(4), 128.1(3), 128.0(4), 127.9(4), 127.8, 127.6, 127.5(2), 127.4, 127.3(2), 127.2, 127.0, 126.5(2), 126.2(2), 123.6, 123.4, 119.2(2), 114.3(2) (ArC), 109.1 [C–(CH3)2], 100.5 (C-1′′), 100.2 (CHPh), 99.5 (C-1′′′), 98.1 (C-1), 97.9 (C-1′′′′′), 97.1 (C-1′), 94.1 (C-1′′′′), 80.4, 80.3, 79.9, 79.1, 78, 75.9, 75.8, 75.5, 75.3, 75.2, 74.8, 73.8, 73.5, 72.8, 72.7, 72.6, 72.3(2), 71.7, 71.1, 71, 70.5, 69.8, 69.2, 68.8, 68.6, 68.3, 66.7, 66.6, 66.3, 62.2, 60.8, 55.5 (C6H4OCH3), 52.3 (C-2), 28.2, 26.2 (2 × isopropylidene-CH3), 20.9, 20.8(2), 20.7 (4 × COCH3), 18.1, 17.8, 16.9 (3 × C–CH3). HRMS calcd for C111H123O34NNa (M + Na)+: 2036.7824, found: 2036.7821.
4-Methoxyphenyl α-L-rhamnopyranosyl (1→4)-β-D-glucopyranosyluronic acid (1→2)-α-L-rhamnopyranosyl-(1→2)-α-L-rhamnopyranosyl-(1→2)-β-D-galactopyranosyl-(1→3)-2-acetamido-2-deoxy-β-D-galactopyranoside (1)
To a solution of the hexasaccharide acceptor 18 (850 mg, 0.4 mmol) in CH2Cl2–H2O (2:1, 20 mL), TEMPO (12 mg, 0.07 mmol) was added followed by iodosobenzene diacetate (IBDA) (310 mg, 1 mmol) and the mixture was vigorously stirred at room temperature for 2 hours till TLC (EtOAc) indicated complete conversion of the starting material to a lower running spot. Na2S2O3 solution (10% in H2O, 10 mL) was added to quench the reaction. The mixture was then extracted with CH2Cl2 (2 × 30 mL). The combined organic layer was dried over Na2SO4, filtered and evaporated in vacuo. The crude acid derivative was then treated with 80% AcOH (10 mL) at 80 °C for 4 hours resulting in the hydrolysis of both benzylidene and isopropylidene ring. The solvents were evaporated and co-evaporated with toluene. The diol formed was dissolved in n-butanol (15 mL) and ethylene diamine (1.5 mL) was added and the reaction mixture was allowed to stir for 24 h at 110 °C. The solvents were then evaporated and co-evaporated with toluene and the crude product thus obtained is dissolved in pyridine (10 mL) followed by the addition of Ac2O (10 mL). The reaction mixture was allowed to stir at room temperature for 10 h when the TLC showed complete conversion of the starting material. The reaction mixture was then co-evaporated with toluene to give the required hexasaccharide, 4-methoxyphenyl-2,3,4-tri-O-acetyl-α-L-rhamnopyranosyl (1→4)-2,3-di-O-benzyl-β-D-glucopyranosyl-(1→2)-3,4-di-O-benzyl-α-L-rhamnopyranosyl-(1→2)-3,4-di-O-benzyl-α-L-rhamnopyranosyl-(1→2)-3,4,6-tri-O-acetyl-β-D-galactopyranosyl-(1→3)-4,6-di-O-acetyl-2-acetamido-2-deoxy-β-D-galactopyranoside in its crude state. The dried compound was dissolved in MeOH (100 mL) and it was passed through an H-cube flow hydrogenation assembly using a 10% Pd–C cartridge with a flow rate of 1 mL min−1. Complete removal of the six benzyl groups was achieved after six cycles as confirmed by the mass spectra. MeOH was evaporated and the compound was dried well. The dried residue was re-dissolved in MeOH (10 mL) and NaOMe (1 mL, 0.5 M in MeOH) was added to it. The reaction mixture was stirred at room temperature for 10 hours. DOWEX 50W H+ resin was added to neutralize excess NaOMe. The solution was filtered, evaporated in vacuo and the residue was triturated with CH2Cl2 to remove the organic soluble side products/salts generated during TEMPO oxidation. The residue obtained thereafter was further purified by HPLC using C18 column and CH3CN–H2O mixture as eluent to afford the desired hexasaccharide 1 (230 mg, 54% overall) as white amorphous mass. [α]25D +61 (c 1.0, MeOH). Partial 1H NMR (500 MHz, CDCl3) δ: 6.98, 6.81 (2d, 4H, J 9.0 Hz, C6H4OCH3), 5.31 (d, 1H, J1,2 8.5 Hz, H-1), 5.06 (d, 1H, J1′′,2′′ 2.0 Hz, H-1′′), 5.01 (d, 1H, J1′,2′ 3.5 Hz, H-1′), 4.93 (s, 1H, H-1′′′′), 4.92 (s, 1H, H-1′′′′′), 4.84 (s, 1H, H-1′′′), 3.73 (s, 1H, C6H4OCH3), 2.02 (s, 3H, NHCOCH3), 1.26 (bd, 9H, J 6.0 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 173.4 (NHCOCH3), 173.0 (COOH), 156.8, 153.2, 119.3(2), 115.5(2) (ArC), 102.3 (C-1′′′), 101.6 (C-1), 101.3 (C-1′′), 101.2 (C-1′′′′′), 99.6 (C-1′′′′), 97.9 (C-1′), 80, 79.4, 78.6, 76.6, 76.2, 76, 74.4, 74.1, 73.8(2), 73.7(2), 72.9, 72.5(2), 72.4, 72.2, 71.6, 71.2, 70.7, 70.6, 70.4, 66.1, 62.6, 62.1, 61.8, 56.1 (C6H4OCH3), 20.9 (NHCOCH3), 18.3, 17.9, 17.8 (3 × C–CH3). HRMS calcd for C45H69O30NNa (M + Na)+: 1126.3802, found: 1126.3798.
Acknowledgements
DB is thankful to UGC, New Delhi for Senior Research Fellowship. This work is funded by the Scientific and Engineering Research Board (SERB), New Delhi through the grant SB/S1/OC-48/2013 to BM.
Notes and references
- P. P. Reeves and L. Wang, Curr. Top. Microbiol. Immunol., 202, 264, 109–135 Search PubMed.
- J. L. Jaegar and D. W. Acheson, Curr. Infect. Dis. Rep., 2000, 2, 61–67 CrossRef.
- P. M. Fratamico, L. K. Bagi, E. J. Bush and B. T. Solow, Appl. Environ. Microbiol., 2004, 70, 7173–7178 CrossRef CAS PubMed.
- M. Blanco, N. L. Padola, A. Kruger, M. E. Sanz, J. E. Blanco, E. A. Gonzalez, G. Dahbi, A. Mora, M. I. Bernardez, A. I. Etcheverria, G. H. Arroyo, P. M. Lucchesi, A. E. Parma and J. Blanco, Int. Microbiol., 2004, 7, 269–276 CAS.
- A. V. Perepelov, Q. Wang, S. N. Senchenkova, Y. Gong, A. S. Shaskov, L. Wang and Y. A. Knirel, Carbohydr. Res., 2012, 353, 106–110 CrossRef CAS PubMed.
- A. K. Choudhury and N. Roy, J. Carbohydr. Chem., 1997, 16, 1363–1371 CrossRef CAS.
- D. Dubreuil, J. Cleophax and A. Loupy, Carbohydr. Res., 1994, 252, 149–157 CrossRef CAS.
- Z. Wang, M. Matin and S. Sheikh, Molecules, 2005, 10, 1325–1334 CrossRef CAS PubMed.
- Z. Zhang and G. Magnusson, Carbohydr. Res., 1996, 295, 41–55 CrossRef CAS PubMed.
-
(a) V. K. Rajput, B. Roy and B. Mukhopadhyay, Tetrahedron Lett., 2006, 47, 6987–6991 CrossRef CAS;
(b) V. K. Rajput and B. Mukhopadhyay, Tetrahedron Lett., 2006, 47, 5939–5941 CrossRef CAS;
(c) B. Mukhopadhyay, Tetrahedron Lett., 2006, 47, 4337–4341 CrossRef CAS;
(d) B. Roy and B. Mukhopadhyay, Tetrahedron Lett., 2007, 48, 3783–3787 CrossRef CAS.
-
(a) S. Dasgupta, B. Roy and B. Mukhopadhyay, Carbohydr. Res., 2006, 341, 2708–2713 CrossRef CAS PubMed;
(b) K. B. Pal and B. Mukhopadhyay, Carbohydr. Res., 2013, 379, 26–29 CrossRef CAS PubMed;
(c) P. R. Verma and B. Mukhopadhyay, RSC Adv., 2013, 3, 201–207 RSC;
(d) R. Das and B. Mukhopadhyay, Carbohydr. Res., 2013, 376, 1–6 CrossRef CAS PubMed;
(e) R. Das, M. Mahanti and B. Mukhopadhyay, Carbohydr. Res., 2014, 399, 15–20 CrossRef CAS PubMed.
- Y. Oikawa, T. Yoshioka and O. Yonemitsu, Tetrahedron Lett., 1982, 23, 885–888 CrossRef CAS.
- T. Ghosh and A. K. Misra, Carbohydr. Res., 2012, 362, 8–12 CrossRef CAS PubMed.
- T. Li, H. Ye, X. Cao, J. Wang, Y. Liu, L. Zhuo, Q. Liu, W. Wang, J. Shen, W. Zhao and P. Wang, ChemMedChem, 2014, 9, 1071–1080 CrossRef CAS PubMed.
- G. Medgyes, G. Jerkovich and J. Kuszmann, Carbohydr. Res., 1989, 186, 225–239 CrossRef CAS PubMed.
- M. Bertolini and C. P. J. Glaudemans, Carbohydr. Res., 1970, 15, 263–270 CrossRef CAS.
- I. Cumpstey, A. J. Fairbanks and A. J. Redgrave, Tetrahedron, 2004, 60, 9061–9074 CrossRef CAS.
- B. Classon, P. J. Garegg and B. Samuelson, Acta Chem. Scand., Ser. B, 1984, 38, 419–422 CrossRef.
- J. Kerékgyártó, Z. Szurmai and A. Lipták, Carbohydr. Res., 1993, 245, 65–80 CrossRef.
- V. K. Rajput and B. Mukhopadhyay, J. Org. Chem., 2008, 73, 6924–6927 CrossRef CAS PubMed.
- K. Takeo, K. Maki, Y. Wada and S. Kitamura, Carbohydr. Res., 1993, 245, 81–96 CrossRef CAS PubMed.
- L. J. van den Bos, J. D. C. Codeé, J. C. van der Toorn, T. J. Boltje, J. H. van Boom, H. S. Overkleeft and G. A. van der Marel, Org. Lett., 2004, 6, 2165–2168 CrossRef CAS PubMed.
- O. Kanie, S. C. Crawley, M. M. Palcic and O. Hindsgaul, Carbohydr. Res., 1993, 243, 139–164 CrossRef CAS PubMed.
- G. Zemplén, Ber. Dtsch. Chem. Ges., 1926, 59, 1254–1266 CrossRef.
- D. D. Perrin, W. L. Amarego and D. R. Perrin, Purification of Laboratory Chemicals, Pergamon, London, 1996 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra of all new compounds. See DOI: 10.1039/c5ra20363e |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence