
Vanadium(V) tetra-phenolate complexes: synthesis, structural studies and ethylene homo-(co-)polymerization capability†
Carl Redshaw*a,
Mark J. Waltonb,
Mark R. J. Elsegoodc,
Timothy J. Priora and
Kenji Michiued
aDepartment of Chemistry, University of Hull, Hull, HU6 7RX, UK. E-mail: c.redshaw@hull.ac.uk
bEnergy Materials Laboratory, School of Chemistry, University of East Anglia, Norwich, NR4 7TJ, UK
cChemistry Department, Loughborough University, Loughborough, Leicestershire LE11 3TU, UK
dProcess Technology Center, Mitsui Chemicals Inc., 580-32 Nagaura, Sodegaura, Chiba 299-0265, Japan
First published on 8th October 2015
Abstract
Reaction of α,α,α′,α′-tetrakis(3,5-di-tert-butyl-2-hydroxyphenyl)-p-xylene (p-L1H4) with two equivalents of [VO(OR)3] (R = nPr, tBu) in refluxing toluene afforded, after work-up, the complexes {[VO(OnPr)(THF)]2(μ-p-L1)}·2(THF) (1·2(THF)) or {[VO(OtBu)]2(μ-p-L1)}·2MeCN (2·2MeCN), respectively in moderate to good yield. A similar reaction using the meta pro-ligand, namely α,α,α′,α′-tetrakis(3,5-di-tert-butyl-2-hydroxyphenyl)-m-xylene (m-L2H4) afforded the complex {[VO(OnPr)(THF)]2(μ-p-L2)} (3). Use of [V(Np-R1C6H4)(tBuO)3] (R1 = Me, CF3) with p-L1H4 led to the isolation of the oxo–imido complexes {[VO(tBuO)][V(Np-R1C6H4) (tBuO)](μ-p-L1)} (R1 = Me, 4·CH2Cl2; CF3, 5·CH2Cl2), whereas use of [V(Np-R1C6H4)Cl3] (R1 = Me, CF3) in combination with Et3N/p-L1H4 or p-L1Na4 afforded the diimido complexes {[V(Np-MeC6H4)(THF)Cl]2(μ-p-L1)}·4toluene (6·4toluene) or {[V(Np-CF3C6H4)(THF)Cl]2(μ-p-L1)} (7). For comparative studies, the complex [(VO)(μ-OnPr)L3]2 (8) has also been prepared via the interaction of [VO(nPrO)3] and 2-(α-(2-hydroxy-3,5-di-tert-butylphenyl)benzyl)-4,6-di-tert-butylphenol (L3H2). The crystal structures of 1·2THF, 2·2MeCN, 3, 4·CH2Cl2, 5·CH2Cl2, 6·4toluene·THF, 7 and 8 have been determined. Complexes 1–3 and 5–8 have been screened as pre-catalysts for the polymerization of ethylene in the presence of a variety of co-catalysts (with and without a re-activator), including DMAC (dimethylaluminium chloride), DEAC (diethylaluminium chloride), EADC (ethylaluminium dichloride) and EASC (ethylaluminium sesquichloride) at various temperatures and for the co-polymerization of ethylene with propylene; results are compared versus the benchmark catalyst [VO(OEt)Cl2]. In some cases, activities as high as 243![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 g mmol−1 V−1 h−1 (30.43 kgPE mmol V−1 h−1 bar−1) were achievable, whilst it also proved possible to obtain higher molecular weight polymers (in comparable yields to the use of [VO(OEt)Cl2]). In all cases with dimethylaluminium chloride (DMAC)/ethyltrichloroacetate (ETA) activation, the activities achieved surpassed those of the benchmark catalyst. In the case of the co-polymerization of ethylene with propylene, complexes 1–3 and 5–8 showed comparable or higher molecular weight than [VO(OEt)Cl2] with comparable catalytic activities or higher in the case of the imido complexes 6 and 7.
400 g mmol−1 V−1 h−1 (30.43 kgPE mmol V−1 h−1 bar−1) were achievable, whilst it also proved possible to obtain higher molecular weight polymers (in comparable yields to the use of [VO(OEt)Cl2]). In all cases with dimethylaluminium chloride (DMAC)/ethyltrichloroacetate (ETA) activation, the activities achieved surpassed those of the benchmark catalyst. In the case of the co-polymerization of ethylene with propylene, complexes 1–3 and 5–8 showed comparable or higher molecular weight than [VO(OEt)Cl2] with comparable catalytic activities or higher in the case of the imido complexes 6 and 7.
Introduction
Interest in the use of group V metal complexes as potential components in catalytic systems for the production of new polymers from α-olefins continues to attract both academic and industrial interest.1 This is in-part driven by the need for new IP in emerging economies such as China and India. In the case of vanadium, interest is further stimulated by the ability to achieve high activities and conduct co- and ter-polymerizations.2 Notable recent successes have been achieved, which have made use of a variety of ligand sets including phenoxyimines, mono-dentate aryloxides, β-enaminoketonato and phenoxy-phosphine/phosphineoxides.3 In our previous work, we have observed very high activities when employing chelating phenoxide ligands, including the use of calix[n]arenes, as well as di-/tri-phenols.4 With this in mind, we were keen to explore other ligand systems that were capable of simultaneously binding to more than one metal centre. A new family of tetraphenols was recently reported by Tang et al.,5 which have since been exploited by the group of Wu to prepare multi alkali-metal complexes capable of the ring opening polymerization (ROP) of L-lactide,6 and by us to afford niobium-based complexes capable of the ROP of ε-caprolactone.7 Herein, we describe the synthesis and molecular structures of a series of vanadyl complexes of this ligand family (shown in Scheme 1), and investigate their polymerization catalysis behaviour towards ethylene and ethylene/propylene under a variety of conditions. Extremely high catalytic activities, of the order of 243400 g mmol−1 V−1 h−1 at 8 bar ethylene (30.43 kgPE mmol V−1 h−1 bar−1), were found to be achievable for these systems which, combined with their ability to afford reasonably high molecular weight products at high temperature, suggests that such systems could be of industrial interest. Indeed, to the best of our knowledge, these are the highest catalytic activities reported to-date for vanadium-based systems for ethylene polymerization under robust conditions. We note though that these high activities, versus related systems, can be ascribed to the use of the high pressures (8 bar) employed herein. For example, the phosphine–phenoxide complex {[2,4-(tBu)2-6-PPh2C6H2O]VCl2(THF)2} can achieve an activity of 41.3 kgPE mmol−1 V−1 h−1 at 1 bar.3d Of the other highly active vanadium systems known (see Scheme 2), the coordination at the metal tends to be a combination of nitrogen (in the form of either an organimido group of an imine linkage) and oxygen (mono- or bi-dentate phenoxide ligation) or, more recently, calix[n]arene derived ligation.3,4 High molecular weight polyethylene is an attractive product given its favorable mechanical and physical properties, though there can be issues with regard to processing.8 We note that industrially, the co-polymerization of ethylene with higher olefins has been successfully achieved by employing group IV-based constrained geometry catalysts. For example, Dow Chemicals has also utilized complexes bearing imino–enamido or pyridyl-amido ligation for ethylene/α-olefin copolymerization and polyolefin block copolymer formation.9,10 However, we note that only a limited number of vanadium-based systems have been reported for ethylene/propylene co-polymerization.4b–d,11
 | ||
| Scheme 1 Pre-catalysts 1–8 prepared herein. | ||
 | ||
| Scheme 2 Known, highly active vanadium-based ethylene polymerization pre-catalysts.3,4 Conditions employed: a[V] = 1.0 μmol, co-cat = MgCl2/EtmAl(OR)n (0.8 mmol Mg:2.4 mmol Et3Al), 75 °C, 1 bar, 15 min. b[V] = 0.8 μmol, co-cat = Me2AlCl (2 mmol), ETA (2 mmol), 80 °C, 7 bar, 15 min. c[V] = 0.05 μmol, co-cat = Et2AlCl (2000 equiv.), 0 °C, 8 bar, 10 min. [V] = 0.1 μmol, co-cat = Et2AlCl (0.05 M), 75 °C, 8 bar, 10 min. d[V] = 0.005 μmol, co-cat = Me2AlCl (20000 equiv.), ETA (20000 equiv.), 80 °C, 8 bar, 30 min. | ||
Results and discussion
Synthesis and structure of p-L1H4 derived vanadyl complexes
The ligand p-L1H4 was synthesized following the reported literature method.5 The compounds {[VO(OnPr)(THF)]2(μ-p-L1)} (1) and {[VO(OtBu)]2(μ-p-L1)} (2) were synthesized in moderate to good yield (45–75%) via the treatment of L1H4 with a slight excess (2.1 equiv.) of [VO(OR)3]. If the reaction is conducted in THF, then this solvent can act as a ligand as in 1, vide infra. Conducting the reaction in toluene and avoiding the use of THF in the work-up affords THF-free products, as in 2. In either case, the reaction proceeds with loss of two equivalents of alcohol per vanadium center. In the case of 1 (R = nPr), crystals suitable for a single crystal X-ray diffraction study were grown by slow diffusion of light petroleum into THF; the crystal structure is presented in Fig. 1 (for ORTEP diagram see Fig. S1 in the ESI†). Each vanadyl center is present in trigonal bipyramidal geometry, and bears an n-propoxide ligand with the fifth position trans to the oxo group occupied by a THF molecule. The two sets of di-phenolates across the central phenyl ring are arranged in a trans fashion related by an inversion center. The bond lengths and angles are given in the caption to Fig. 1, and are typical and similar to the other vanadium complexes in trigonal bipyramidal geometry.12 An 8-membered metallocycle is formed at each end of the tetra-phenolate, with each adopting the chair-boat conformation. The bite angle of the chelate is 111.73(7)°, which is somewhat larger than that found in the mononuclear vanadyl complex {VOCl[2,2′-CH2(4-Me-6-tBuC6H2O)2]} (106.9(2)°) and the dimeric complex {VO(OnPr)(2,2′-CH3CH[4,6-(tBu)2C6H2)2]} (94.49(10)°).4,13 In the IR spectra for 1 and 2, a strong band at ca. 990 cm−1 is assigned to the ν(V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) mode.
O) mode.
 | ||
| Fig. 1 Representation of the centrosymmetric molecular structure of complex 1 in the solid state, indicating the atom numbering scheme. tert-Butyl groups, hydrogen atoms, and unbound solvent molecules have been removed for clarity. Selected bond lengths (Å) and angles (°): V1–O3 1.5871(14), V1–O5 1.7878(15), V1–O2 1.8187(15), V1–O1 1.8256(16), V1–O4 2.3307(13), O3–V1–O5 100.20(7), O3–V1–O2 100.05(7), O5–V1–O2 119.29(7), O3–V1–O1 99.63(7), O5–V1–O1 120.16(7), O2–V1–O1 111.73(7), O3–V1–O4 178.12(7), O5–V1–O4 78.36(6), O2–V1–O4 81.73(6), O1–V1–O4 80.16(6). Symmetry operation used to generate equivalent atoms: i = 1−x, −y, −z. | ||
In complex 2, each vanadyl centre is bound by the bi-dentate di-phenolate, forming an 8-membered metallocycle with a bite angle at the metal of 112.0(2)°. The coordination is completed by a single tert-butoxide ligand to form a slightly distorted tetrahedral geometry. The complex is centrosymmetric (Fig. 2; for ORTEP diagram see Fig. S2 in the ESI†) with the two vanadyl cations lying on opposite sides of the plane of the central phenyl ring in similar fashion to 1.
 | ||
| Fig. 2 Representation of the centrosymmetric molecular structure of complex 2 in the solid state, indicating the atom numbering scheme. Hydrogen atoms, and unbound solvent molecules have been removed for clarity. Selected bond lengths (Å) and angles (°): O1–V1 1.725(6), O2–V1 1.786(5), O3–V1 1.547(6), O4–V1 1.682(7), O3–V1–O4 112.4(4), O3–V1–O1 106.8(3), O4–V1–O1 110.1(3), O3–V1–O2 106.1(3), O4–V1–O2 109.3(3), O1–V1–O2 112.0(2). Symmetry operation used to generate equivalent atoms: i = 1−x, 1−y, −z. | ||
Synthesis and structure of m-L2H4 derived vanadyl complexes
Similar treatment of the meta ligand L2H4 with a slight excess of [VO(OnPr)3] led to the formation of the complex {[VO(OnPr)]2(μ-m-L2)} (R = nPr (3)). The IR spectrum contains a strong band at ca 990 cm−1 assigned to the ν(VO) mode. Unsurprisingly, the 51V NMR spectrum is very similar to that of 1 with a single peak at δ −432.5 with ω1/2 170 Hz (cf. −433.3 ppm, ω1/2 170 Hz for 1); see Table S5† for all 51V NMR spectroscopic data. Crystals of 3 suitable for an X-ray diffraction study were obtained on cooling of a THF/light petroleum solution to −20 °C. A tiny orange platelet was extracted from the solid product. This was examined at 100 K using synchrotron radiation (DLS beam-line I19, λ = 0.6889 Å).14 The crystal was extremely weakly scattering and no significant diffraction was obvious beyond ca. 2θ = 36°. It proved possible to solve the structure using direct methods and the chemical connectively is unequivocally established (Fig. 3; for ORTEP diagram see Fig. S3 in the ESI†). No data beyond 36° were used in the structure refinement.
 | ||
| Fig. 3 One of the symmetry unique complexes within 3 showing the meta arrangement in ligand L2. Tertiary butyl groups and hydrogen atoms have been omitted for clarity. Symmetry equivalent atoms are generated by the operator i = 1/2−x, y, 1/2−z. | ||
The structure contains two independent units that are chemically identical. Each is formed of one half of a molecule of L2. The second half is generated by 2-fold rotation symmetry. Each symmetry unique fragment is based upon bidentate coordination of L2 to the vanadyl cation. Coordination about the vanadyl is completed by tetrahydrofuran and n-propoxide to give trigonal bipyramidal geometry about the V center. There is some evidence for disordered solvent between these coordination complexes but we have not been able to resolve this.
Synthesis and structure of oxo–imido complexes
The effective use of (imido)vanadium complexes as pre-catalysts for α-olefin polymerization has been noted previously.15 Given this, we have also explored possible routes to accessing imido-containing vanadium complexes of the tetra-phenol ligand set.Treatment of the para ligand L1H4 with a slight excess of [V(Np-R1C6H4)(OtBu)3] (R1 = Me, CF3) led to the formation of the oxo–imido complexes {[VO(tBuO)][V(Np-R1C6H4)(tBuO)](μ-p-L1)} (R1 = Me, 4; CF3, 5). Crystals of 4 and 5 suitable for X-ray diffraction studies were obtained on prolonged cooling of saturated dichloromethane solutions (to −20 °C). The molecular structures are shown in Fig. 4 and 5 (for ORTEP diagrams see Fig. S4 and S5 in the ESI†), with selected bond lengths (Å) and angles (°) given in Table 1. For 4, each vanadium center adopts a pseudo-tetrahedral geometry, with bond angles in the range 106.7(2)–112.1(2)°. Each 8-membered metallocycle adopts the boat conformation, and the bite angle of the chelate at each end is 111.80(18)°. The tert-butoxide ligand is again somewhat bent [V1–O4–C40 = 144.5(4)°], whilst the organoimido group is near linear [V1–N1–C45 = 172.8(6)°].
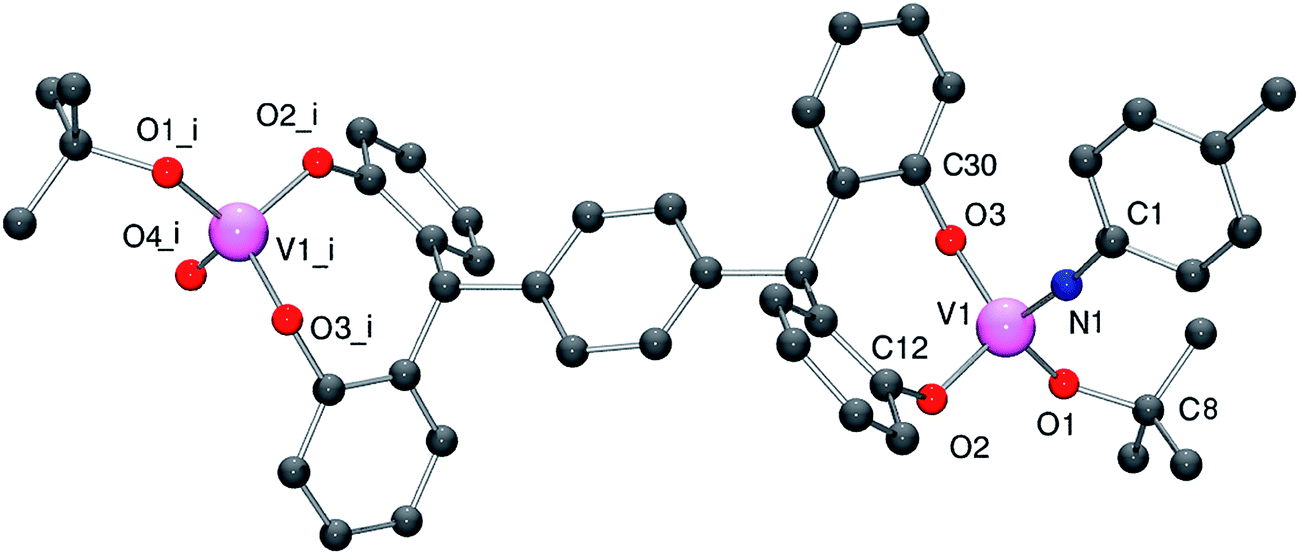 | ||
| Fig. 4 Molecular structure of complex 4·2CH2Cl2, indicating the atom numbering scheme. tert-Butyl groups, hydrogen atoms, and unbound solvent molecules have been removed for clarity. Symmetry equivalent atoms are generated by the operator i = 1−x, 1−y, 2−z. | ||
 | ||
| Fig. 5 Molecular structure of complex 5·2CH2Cl2, indicating the atom numbering scheme. tert-Butyl groups, hydrogen atoms, and unbound solvent molecules have been removed for clarity. Symmetry operation used to generate equivalent atoms: i = 1−x, 1−y, 1−z. | ||
| Bond lengths (Å)/angles (°) | 4·2CH2Cl2 | 5·2CH2Cl2 |
|---|---|---|
| V1–N1 | 1.600(5) | 1.603(5) |
| V1–O1 | 1.735(4) | 1.734(5) |
| V1–O2 | 1.805(4) | 1.803(5) |
| V1–O3 | 1.797(4) | 1.802(4) |
| V1–O1–C8 | 144.4(4) | 144.3(5) |
| V1–O2–C12 | 125.7(4) | 126.0(4) |
| V1–O3–C30 | 127.5(4) | 125.2(4) |
| O1–V1–N1 | 112.1(2) | 111.8(3) |
In the case of 5, as in 4, the molecule also lies on a center of symmetry and so again half is unique. There is therefore disorder such that at V1 there is a 50/50 mixture of (i) a vanadyl and an unbound CH2Cl2 molecule and (ii) the p-arylimido group. We interpret this as being (i) at one end and (ii) at the other but that the arrangement is not regular throughout the crystal. The structure of 5 is almost isomorphous with 4. As in 4, the organoimido group is near linear [V1–N1–C1 = 173.4(6)°], whereas the alkoxide is bent [V1–O1–C8 144.3(5)°]. The vanadyl group is involved in H-bonding to a solvent molecule (CH2Cl2) with the geometrical parameters H45A⋯O4 = 2.20 Å, angle at H45 = 167°, H45B⋯centroid of aromatic ring C30 to C35 = 2.85 Å, angle at H45B = 136°. Molecules of 5 pack into chains but there are no significant interactions between molecules and chains.
Synthesis and structure of bis-imido complexes
Given the air sensitive nature of the tert-butoxides employed above, we turned out attention to use of the parent trichlorides, namely [V(Np-R1C6H4)Cl3].16Interaction of [V(Np-MeC6H4)Cl3] with the sodium salt p-L1Na4 afforded the complex {[V(Np-MeC6H4)(THF)Cl]2(μ-p-L1)} (6·4toluene) as a red/brown crystalline solid. The molecular structure of 6·4toluene is shown in Fig. 6 (for ORTEP diagram see Fig. S6 in the ESI†), with selected bond lengths and angles given in the caption. The geometry at vanadium is best described as trigonal bipyramidal with the imido and THF groups occupying axial positions [N(1)–V(1)–O(3) 175.11(16)°]. Distortions are in the range 110.17(12)–123.9(2)°, with the largest deviation associated with the angle subtended at the metal by the phenolic oxygen centers; the metallocycle adopts a boat conformation. The imido ligand has the geometrical parameters associated with a linear imido function [V(1)–N(1) 1.656(4) Å; V(1)–N(1)–C(50) 170.4(3)°].
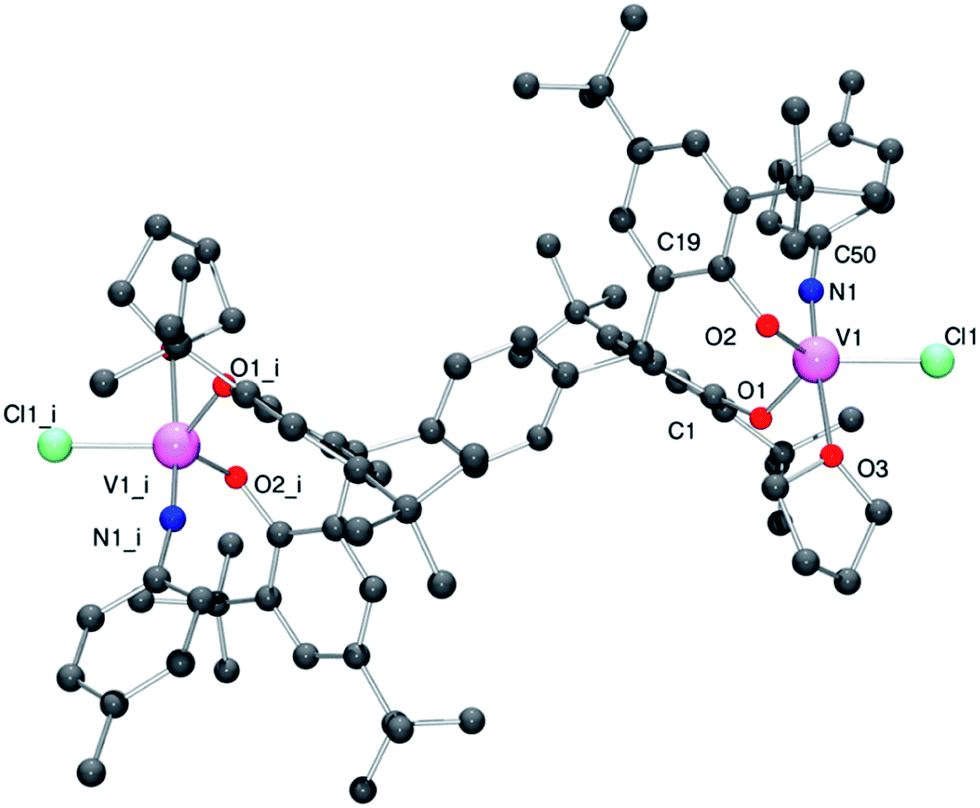 | ||
| Fig. 6 Molecular structure of complex 6·4toluene, indicating the atom numbering scheme. Hydrogen atoms have been removed for clarity. Selected bond lengths (Å) and angles (°): V(1)–N(1) 1.656(4), V1–O1 1.819(3), V1–O2 1.817(3), V1–O3 2.189(3), V1–Cl1 2.2814(14); O1–V1–N1 98.84(15), O1–V1–O2 110.17(12), Cl1–V1–N1 92.89(12), V1–O1–C1 126.9(2), V1–O2–C19 120.6(3), V1–N1–C50 170.4(3). Symmetry operation used to generate equivalent atoms: i = −x, 1−y, −z. | ||
Similar use of [V(Np-CF3C6H4)Cl3] with L1H4 led to the isolation of {[V(Np-CF3C6H4) (THF)Cl]2(μ-p-L1)} (7) in moderate yield (ca. 47%). Crystals suitable for X-ray diffraction were obtained from a saturated solution of acetonitrile at ambient temperature. Although the data are not of the best quality, the connectivity is clear and the molecular structure is shown in Fig. 7 (for ORTEP diagram see Fig. S7 in the ESI†). In the 51V NMR spectra of 4 and 5 there are two peaks (see Table S5†); imido peaks are usually found downfield of their vanadyl counterparts.16
 | ||
| Fig. 7 Molecular structure of complex 7, indicating the atom numbering scheme. Hydrogen atoms and tert-butyl groups have been removed for clarity. Selected bond lengths (Å) and angles (°): V(1)–N(1) 1.666(4), V1–O1 1.808(3), V1–O2 1.819(3), V1–O3 2.210(3), V1–Cl1 2.2625(14); O1–V1–N1 99.05(18), O1–V1–O2 112.47(14), Cl1–V1–N1 95.29(14), V1–O1–C1 122.7(3), V1–O2–C19 121.5(3), V1–N1–C34 172.0(4). | ||
Under the conditions employed herein, on several occasions, small amounts of spiro-type compounds containing the motif I (see Fig. 8) were isolated. In particular, the spiro compound with X = tBu was isolated from the reaction employing [V(NC6H4CF3-p)Cl3], whilst that with X = Cl resulted from attempts to form a vanadyl chloride complex of p-L1H4 using [VOCl3]. The crystal structures of both spiro compounds are presented in the ESI (see Fig. S8 and S9 and Tables S1–S4†). A search of the Cambridge Crystallographic Database (CSD) for motifs related to I revealed 7 hits in calixarene type systems.17,18
 | ||
| Fig. 8 Spiro motif I. | ||
Use of L3H2
For comparative studies, we have also treated the potentially bidentate ligand L3H2 with [VO(OnPr)3], which led to the formation of compound 8. In the IR spectrum, a strong band at 989 cm−1 is assigned to the vanadyl group. Compound 8 was crystallized from light petroleum to give red needles which were suitable for single crystal X-ray diffraction. The crystal structure revealed that compound 8 forms a dimeric structure in the solid state (see Fig. 9; for ORTEP diagram see Fig. S10 in the ESI†). The vanadium oxytri-n-propoxide loses two equivalents of propanol on binding to the bidentate ligand. The dimer is centrosymmetric and contains two vanadyl moieties in a trans arrangement that are bridged by the two remaining n-propoxide ligands. Each vanadium metal center is in trigonal bipyramidal geometry; the bidentate ligand and one of the n-propoxide ligands occupying the equatorial position, the vanadyl oxygen and second n-propoxide occupies the axial position. The di-phenolate ligand's third phenol ring is rotated away with the methine hydrogen directed toward the vanadium center. Unlike for complexes 1 and 3 which contain terminal n-propoxide ligands, the presence of the bridging n-propoxides in 8 together with the bulky chelate ligand appears to prevent ligation by THF. As in the tetra-phenolate systems above, the 8-membered metallocycle in 8 adopts a boat conformation, for which the bite angle subtended at vanadium is 113.14(7)°. A search of the CSD for use of di-phenols with an aryl group bound at the bridging carbon afforded 114 hits for metal complexes, however most of these were either based on tripodal ligands or where the motif of interest formed part of a more exotic ligating species. Indeed, there was only one example of the previous use of the parent L3H2, which was a report of its utilization in titanium chemistry.17,19 | ||
| Fig. 9 Representation of the molecular structure of complex 8, indicating the atom numbering scheme. For clarity, hydrogen atoms have been removed. One of the symmetry independent t-butyl groups is disordered over two positions. Selected bond lengths (Å) and angles (°): O1–V1 1.8117(15), O2–V1 1.8165(16), O3–V1 1.5869(15), O4–V1i 1.8348(15), O4–V1 2.2917(14), V1–O4i 1.8348(15), V1i–O4–V1 108.23(6), O3–V1–O1 100.19(7), O3–V1–O2 100.58(8), O1–V1–O2 113.14(7), O3–V1–O4i 100.35(7), O1–V1–O4i 115.74(7), O2–V1–O4i 121.56(7), O3–V1–O4 172.06(7), O1–V1–O4 84.55(6), O2–V1–O4 83.19(6), O4i–V1–O4 71.77(6). Symmetry equivalent atoms are generated by the operator i = 1−x, 1−y, 2−z. | ||
Ethylene polymerization screening
Compounds 1–3, 5–8 and [VO(OEt)Cl2], were screened for the polymerization of ethylene. Each catalyst has been screened for polymerization using different co-catalysts (DMAC, dimethylaluminium chloride; DEAC, diethylaluminium chloride; EADC, ethylaluminium dichloride; EASC, ethylaluminium sesquichloride) and with addition of ETA (ethyltrichloroacetate). From the co-catalyst screening (Tables 2, S6, S7 and Fig. 10 and S11–S46 in the ESI;† for use of Me3Al, Et3Al and DMAO (dried MAO – see general experimental), see Tables S8 and S9, ESI†), the addition of ETA to the catalytic system is beneficial; the activity of the runs including an addition of ETA was always higher than with no addition (Table 2).20 In all cases, the addition of larger equivalence of ETA and co-catalyst lead to improved activity. Use of different chloro-aluminium alkyls indicated, for compound 1, that DEAC was the co-catalyst of choice giving the highest activity and lowest molecular weight distribution (Table 2, run 4); compound 3 gave similar activities for each co-catalyst (i.e. no advantage of meta versus para ligation), whereas surprisingly, given the similarities with 3, compound 8 gave much higher activities when using the ethyl derived aluminium chloride co-catalysts (see Table 2, runs 20–27). For compounds 1 and 3, EADC and EASC gave lower activities than DEAC and lower molecular weights than DMAC. The highest molecular weight polyethylene was obtained using DMAC as co-catalyst; however the PDI values were high for each pre-catalyst employed suggesting multiple active species.| Run | Pre-Cat | Co-Cat | Al/V | ETA/V | Tc | Yieldb | Activityd | Mw | Mn | PDI |
|---|---|---|---|---|---|---|---|---|---|---|
| a Conditions: 50 °C, 5 mL toluene, 0.01 μmol V, 0.8 MPa ethylene, reaction quenched with isobutyl alcohol.b Grams.c Minutes.d (g mmol−1 V−1 h−1).e Polymerization was stopped due to consumption of stock ethylene. See ESI for full screening results – Table S5. | ||||||||||
| 1 | 1 | DMAC | 20000 |
30 | 0.128 | 12800 |
2261718 |
547734 |
4.1 | |
| 2 | 20000 |
20000 |
30 | 0.534 | 53400 |
974413 |
116671 |
8.4 | ||
| 3 | DEAC | 20000 |
30 | 0.009 | 900 | 455970 |
88930 |
5.1 | ||
| 4 | 20000 |
20000 |
10e | 0.811 | 243400 |
73074 |
25671 |
2.9 | ||
| 5 | EADC | 20000 |
30 | 0.05 | 5000 | 228678 |
94326 |
2.4 | ||
| 6 | 20000 |
20000 |
30 | 0.338 | 33800 |
749498 |
205539 |
3.7 | ||
| 7 | EASC | 20000 |
30 | 0.058 | 5800 | 943144 |
353483 |
2.7 | ||
| 8 | 20000 |
20000 |
30 | 0.358 | 35800 |
666983 |
160727 |
4.2 | ||
| 9 | 2 | DMAC | 20000 |
20000 |
30 | 0.306 | 122200 |
1180000 |
241000 |
4.9 |
| 10 | DEAC | 20000 |
20000 |
30 | 0.231 | 92300 |
1040000 |
103000 |
10.0 | |
| 11 | 3 | DMAC | 20000 |
30 | 0.112 | 11200 |
3290580 |
1349253 |
2.44 | |
| 12 | 20000 |
20000 |
30 | 0.45 | 45000 |
865647 |
81817 |
10.6 | ||
| 13 | DEAC | 20000 |
20000 |
30 | 0.494 | 49400 |
267660 |
45443 |
5.9 | |
| 14 | EADC | 20000 |
30 | 0.01 | 1000 | 196554 |
57342 |
3.4 | ||
| 15 | 20000 |
20000 |
30 | 0.472 | 47200 |
749897 |
70411 |
10.6 | ||
| 16 | EASC | 20000 |
30 | 0.034 | 3400 | 967997 |
315747 |
3.1 | ||
| 17 | 20000 |
20000 |
30 | 0.438 | 43800 |
273709 |
27818 |
9.8 | ||
| 18 | 5 | DMAC | 20000 |
20000 |
30 | 0.256 | 102200 |
814000 |
123000 |
6.6 |
| 19 | DEAC | 20000 |
20000 |
30 | 0.237 | 94700 |
933000 |
88800 |
10.5 | |
| 20 | 8 | DMAC | 20000 |
30 | 0.341 | 34100 |
1683732 |
57625 |
29.2 | |
| 21 | 20000 |
20000 |
30 | 0.423 | 42300 |
1067563 |
120753 |
8.84 | ||
| 22 | DEAC | 20000 |
30 | 0.039 | 3900 | 333763 |
104963 |
3.2 | ||
| 23 | 20000 |
20000 |
15e | 0.527 | 105400 |
110765 |
26097 |
4.2 | ||
| 24 | EADC | 20000 |
30 | 0.0107 | 1100 | 181151 |
68551 |
2.6 | ||
| 25 | 20000 |
20000 |
23e | 0.538 | 70200 |
547756 |
135546 |
4.0 | ||
| 26 | EASC | 20000 |
30 | 0.0305 | 3100 | 874365 |
191854 |
4.6 | ||
| 27 | 20000 |
20000 |
22e | 0.4951 | 67500 |
216581 |
29627 |
7.3 | ||
| 28 | [VO(OEt)Cl2] | DMAC | 20000 |
30 | 0.106 | 21200 |
2114696 |
740175 |
2.9 | |
| 29 | 20000 |
20000 |
30 | 0.429 | 85800 |
1787338 |
385401 |
4.6 | ||
| 30 | DEAC | 20000 |
20000 |
30 | 0.343 | 68600 |
139330 |
48676 |
2.9 | |
| 31 | EADC | 20000 |
20000 |
30 | 0.375 | 37500 |
711232 |
187694 |
3.8 | |
| 32 | EASC | 20000 |
20000 |
30 | 0.341 | 34100 |
387638 |
99975 |
3.9 | |
Comparison of the observed catalytic activities and molecular weights (Mw) for the dinuclear vanadyl complexes bridged with a tetra-phenolate ligand versus the n-propoxide bridged vanadyl complex 8 revealed that the use of different co-catalysts as well as the presence or absence of ETA played a significant role. For example, in the presence of ETA, all tetra-phenolate bridged systems were more active than the n-propoxide bridged vanadyl complex 8, however the polyethylene produced via 8/ETA tended to be of higher molecular weight (Mw). In the absence of ETA, 8/DMAC exhibited higher activities than did the tetra-phenolate bridged systems, although in this case, the polyethylene was of higher molecular weight for the tetra-phenolate bridged systems. The use of DEAC as co-catalyst generally afforded the same trends as DMAC for 1, whereas for 2, 3 and 5 with ETA, the activities were less than observed for 8/ETA; molecular weights were higher for the tetra-phenolate bridged systems. With EADC or EASC, activities in the presence of ETA were higher for 8, but molecular weights for the PE obtained were higher for the tetra-phenolate bridged systems. In the absence of ETA, activities for 1 were higher than for 8, whilst for 3 they were about the same as those observed for 8; molecular weights for the PE obtained were higher for the tetra-phenolate bridged systems.
Using the conditions established in Table 2 (20000 equivalence DEAC or DMAC, 20000 equivalence ETA) compounds 1–3, 5–8 and the reference compound [VO(OEt)Cl2] were screened over a series of temperatures (Tables 3 and S7 and Fig. 10 and S27–S42 in the ESI†). When DMAC was used as co-catalyst, the vanadyl-containing pre-catalysts 1–3, 5 and [VO(OEt)Cl2] showed optimal activity at 50 °C, whereas the n-propoxide bridged pre-catalyst 8 is thermally more stable and gave highest activity at 80 °C; each compound except for compound 8 showed lower PDI values at 80 °C. In the runs where DEAC was employed as co-catalyst again 50 °C was the temperature of choice, except for compound 1 where a temperature of 80 °C showed increased activity.
| Run | Pre-Cat | Co-Cat | Tempc | Td | Yieldb | Activitye | Mw | Mn | PDI | Tmc |
|---|---|---|---|---|---|---|---|---|---|---|
| a Conditions: 5 mL toluene, 0.005 μmol V, 0.8 MPa ethylene, 20000 equivalents co-catalyst, 20000 equivalents ETA, reaction quenched with iso-butyl alcohol.b Grams.c °C.d Minutes.e (g mmol−1 V−1 h−1). |
||||||||||
| 1 | 1 | DMAC | 50 | 30 | 0.422 | 168800 |
467300 |
67300 |
6.9 | 137.2 |
| 2 | 80 | 30 | 0.41 | 163900 |
136100 |
44500 |
3.1 | 133.2 | ||
| 3 | DEAC | 50 | 30 | 0.283 | 113200 |
254000 |
38900 |
6.5 | 135.4 | |
| 4 | 80 | 30 | 0.355 | 148000 |
135800 |
20600 |
6.7 | 136.1 | ||
| 5 | 2 | DMAC | 50 | 30 | 0.306 | 122200 |
1180000 |
241000 |
4.9 | 136.1 |
| 6 | 80 | 30 | 0.217 | 86800 |
249000 |
26800 |
9.3 | 136.0 | ||
| 7 | DEAC | 50 | 30 | 0.231 | 92300 |
1040000 |
103000 |
10.0 | 131.8 | |
| 8 | 80 | 30 | 0.110 | 44100 |
82000 |
18800 |
4.4 | 134.3 | ||
| 9 | 3 | DMAC | 50 | 30 | 0.357 | 142800 |
536200 |
93900 |
5.7 | 132.0 |
| 10 | 80 | 30 | 0.301 | 120600 |
173800 |
60400 |
2.9 | 133.0 | ||
| 11 | DEAC | 50 | 30 | 0.218 | 87400 |
555100 |
84000 |
6.6 | 134.0 | |
| 12 | 80 | 30 | 0.046 | 18400 |
366500 |
34900 |
10.5 | 133.0 | ||
| 13 | 5 | DMAC | 50 | 30 | 0.256 | 102200 |
814000 |
123000 |
6.6 | 131.9 |
| 14 | 80 | 30 | 0.224 | 89700 |
163000 |
44800 |
3.6 | 133.5 | ||
| 15 | DEAC | 50 | 30 | 0.237 | 94700 |
933000 |
88800 |
10.5 | 132.2 | |
| 16 | 80 | 30 | 0.040 | 16200 |
142000 |
16100 |
8.8 | 133.1 | ||
| 17 | 6 | DMAC | 50 | 30 | 0.3099 | 124000 |
1430000 |
265000 |
5.4 | 136.8 |
| 18 | 80 | 30 | 0.034 | 13600 |
119000 |
30800 |
3.8 | 134.3 | ||
| 19 | DEAC | 50 | 30 | 0.5899 | 236000 |
162000 |
62800 |
2.6 | 135.1 | |
| 20 | 80 | 30 | 0.1439 | 57600 |
65800 |
35000 |
1.9 | 134.1 | ||
| 21 | 7 | DMAC | 50 | 30 | 119.84 | 119800 |
996000 |
132000 |
7.5 | 134.3 |
| 22 | 80 | 30 | 32.56 | 32600 |
131000 |
52800 |
2.5 | 133.7 | ||
| 23 | DEAC | 50 | 30 | 0.4856 | 194200 |
173000 |
58600 |
3.0 | 134.6 | |
| 24 | 80 | 30 | 0.0709 | 28400 |
72400 |
39500 |
1.8 | 133.6 | ||
| 25 | 8 | DMAC | 50 | 30 | 0.311 | 124400 |
783800 |
115000 |
6.8 | |
| 26 | 80 | 30 | 0.402 | 161000 |
188000 |
67800 |
2.8 | |||
| 27 | DEAC | 50 | 30 | 0.215 | 86100 |
671700 |
88100 |
7.6 | ||
| 28 | 80 | 30 | 0.056 | 22600 |
313500 |
35800 |
8.8 | |||
| 29 | [VO(OEt)Cl2] | DMAC | 50 | 30 | 0.374 | 74700 |
945800 |
168700 |
5.6 | 134.7 |
| 30 | 80 | 30 | 0.354 | 70800 |
137600 |
45700 |
3.0 | 133.3 | ||
| 31 | DEAC | 50 | 30 | 0.483 | 96700 |
316500 |
56800 |
5.6 | 134.4 | |
| 32 | 80 | 30 | 0.237 | 47400 |
208700 |
27200 |
7.7 | 134.0 | ||
For the non-vanadyl (imido) pre-catalysts 6 and 7, observed activities were higher when employing DEAC versus DMAC at 50 °C, with activities as high as 236000 g mmol−1 V−1 h−1 (29.5 kgPE mmol V−1 h−1 bar−1) recorded; pre-catalyst 6 bearing the p-tolyl groups gave higher activities than 7 bearing the p-CF3 group at 50 °C for both DMAC and DEAC. At 80 °C, the activities fell off dramatically when using either DMAC or DEAC, with no products isolated at the higher temperatures of 100 and 144 °C. Indeed, the fall-off in the observed activity was far steeper for these imido systems than was observed for any of the vanadyl systems. The molecular weights of the polymers isolated at 50 and 80 °C were higher when obtained in the presence of DMAC, and in the case of 6 at 50 °C, were larger than any of the molecular weights (Mw) observed herein when using the vanadyl complexes. However, the molecular weights (Mw) of the PE obtained using 6 or 7 in combination with DEAC/ETA were much lower than those observed when employing the vanadyl complexes under similar conditions.
The polyethylene formed is highly linear, with melting points in the range 130.2–137.2 °C, and no branching could be assigned from the 13C NMR spectra (for example, see Fig. S43 in the ESI for catalyst system using 2/Et3AlCl2 – run 31, Table S6†).21
Screening using Me3Al, Et3Al or DMAO as co-catalysts in the presence of ETA proved unsuccessful (see Tables S8 and S9, ESI†), i.e. such systems were inactive under the conditions employed herein. We note that improved activities in vanadium-based systems in the presence of chloroaluminium co-catalysts have previously been associated with the presence of V–Cl–Al type motifs present in the active species,2b and with the nature of the ion-pair formed. Smaller co-catalysts such as DMAC or DEAC versus MAO are capable of equilibria involving chloro-bridged species and discrete ions.22
Ethylene/propylene co-polymerization screening
The co-polymerization of propylene and ethylene using compounds 1–3, 5–8 and [VO(OEt)Cl2] at 50 °C revealed (see Table 4 and Fig. 11 and S44–S46 in the ESI†) that DMAC, in combination with the vanadyl-containing pre-catalysts, was a more efficient co-catalyst than DEAC, achieving an activity greater than 100000 g mmol−1 V−1 h−1 for pre-catalysts 1, 3, 8 and [VO(OEt)Cl2]. As for the homo-polymerization of ethylene, there was no advantage observed for the co-polymerization results herein when using a meta ligand framework over para ligation (1 vs. 3), despite the increased possibility of the former to bring the metals into closer proximity.
| Run | Pre-Cat | Co-Cat | Yieldb | Activityc | %C3d | Mw | Mn | PDI | Tme |
|---|---|---|---|---|---|---|---|---|---|
| a Conditions: 5 mL toluene, 30 minutes, 50 °C, 0.005 μmol V, 0.4 MPa ethylene, 0.4 MPa propylene, 20000 equivalents co-catalyst, 20000 equivalents ETA, reaction quenched with isobutyl alcohol.b Grams.c (g mmol−1 V−1 h−1).d Mol% determined by IR.e °C. |
|||||||||
| 1 | 1 | DMAC | 0.361 | 144400 |
8.5 | 325200 |
133600 |
2.4 | 90.4 |
| 2 | DEAC | 0.203 | 81000 |
8.3 | 88800 |
46400 |
1.9 | 93.4 | |
| 3 | 2 | DMAC | 0.145 | 57880 |
8.6 | 217100 |
100500 |
2.2 | 86.6 |
| 4 | DEAC | 0.084 | 33480 |
7.2 | 78000 |
40300 |
1.9 | 91.6 | |
| 5 | 3 | DMAC | 0.338 | 135100 |
8.2 | 291100 |
123100 |
2.4 | 90.9 |
| 6 | DEAC | 0.116 | 46400 |
7.6 | 98300 |
43200 |
2.3 | 95.0 | |
| 7 | 5 | DMAC | 0.214 | 85560 |
7.5 | 241200 |
103800 |
2.3 | 89.1 |
| 8 | DEAC | 0.171 | 68240 |
8.5 | 87200 |
42800 |
8.5 | 89.9 | |
| 9 | 6 | DMAC | 0.082 | 32960 |
4.7 | 301900 |
123900 |
2.4 | 89.6 |
| 10 | DEAC | 0.278 | 111360 |
7.4 | 108000 |
53700 |
2.0 | 88.2 | |
| 11 | 7 | DMAC | 0.099 | 39440 |
3.8 | 289500 |
117800 |
2.5 | 86.8 |
| 12 | DEAC | 0.199 | 79760 |
4.0 | 102400 |
53800 |
1.9 | 84.5 | |
| 13 | 8 | DMAC | 0.274 | 109600 |
8.2 | 311300 |
136900 |
2.3 | 90.9 |
| 14 | DEAC | 0.189 | 75600 |
7.7 | 99300 |
53000 |
1.9 | 93.6 | |
| 15 | [VO(OEt)Cl2] | DMAC | 0.391 | 156200 |
10.0 | 241100 |
86600 |
2.8 | 88.9 |
| 16 | DEAC | 0.191 | 76400 |
9.1 | 75700 |
42700 |
1.8 | 90.2 | |
 | ||
| Fig. 10 Activity (×103 g mmol−1 V−1 h−1) in ethylene polymerization at 50–140 °C by [VO(OEt)Cl2], 1–3, 5–8. | ||
 | ||
| Fig. 11 Activity (×103 g mmol−1 V−1 h−1) in ethylene/propylene co-polymerization at 50 °C by [VO(OEt)Cl2], 1–3 and 5–8 in the presence of DMAC or DEAC as co-catalyst. | ||
For the non-vanadyl systems 6 and 7, the activity observed was higher when using DEAC as co-catalyst, with the system employing pre-catalyst 6 achieving an activity of the order of 111400 g mmol−1 V−1 h−1. In all cases, the molecular weight (Mw) of the co-polymer produced was much higher when DMAC was employed as co-catalyst. In each run, the PDI values were typically in the range 1.8–2.8 (the exception was run 8). When using DMAC, the propylene incorporation for the vanadyl-containing systems was between 7.5–8.6% (7.2–8.5% for DEAC), whereas for the imido-containing systems, the incorporation was somewhat lower at 3.8–4.7% (DMAC) and 4.0–7.4% (DEAC). For the n-propoxide complex 8, propylene incorporation was similar to the other vanadyl complexes [8.2% DMAC and 7.7% DEAC], whilst the C3 incorporation for the standard catalyst [VO(OEt)Cl2] was slightly higher at 10.0% (DMAC) and 9.1% (DEAC).
Whilst the catalytic activities of the systems described herein (see Fig. 11) are amongst the highest yet reported for ethylene/propylene co-polymerization for vanadium-based systems, the degree of propylene incorporation [3.8–8.6 mol%] is far lower than other reported systems; typically other systems incorporate between 15–40 mol% C3.4b–d,11
In conclusion, the para or meta-tetra-phenols p-L1H4 or m-L2H4 on reaction with [V(X)(OR)3] (X = oxo or imido) allow access to two new families of bimetallic complexes capable of ethylene polymerization with high activity bearing either two vanadyl centers or a combination of a vanadyl/vanadium imido center. Access to bimetallic complexes possessing two vanadium imido centers was achieved using the imido trichloride precursors of the type [V(NAr)Cl3] (Ar = p-tolyl, p-CF3C6H4) via either salt metathesis or HCl elimination.
The tetra-phenolate vanadyl complexes 1–3 and 5, when activated with DMAC or DEAC/ETA, showed higher activities than the benchmark catalyst [VO(OEt)Cl2] for ethylene polymerization; better performances were observed in the presence of DMAC. No advantages were observed when employing meta versus the para ligation. The non-vanadyl (imido) complexes 6 and 7 performed better in the presence of DEAC and at 50 °C achieved activities as high as 230000 g mmol−1 V−1 h−1, which is the highest activity reported to-date under such robust conditions. In the case of ethylene/propylene co-polymerization, the complexes described herein gave higher molecular weight copolymer than did [VO(OEt)Cl2] at comparable activity. Large variation of catalytic activity and polymer (or co-polymer) molecular weight (Mw) was observed on variation of the co-catalyst employed and whether the re-activator ETA was present or not for the various types of vanadium pre-catalyst deployed herein. The extremely high activities observed for these vanadium-based systems suggest that they are of potential industrial use.
Experimental
General
All manipulations were carried out under an atmosphere of dry nitrogen using conventional Schlenk and cannula techniques or in a conventional nitrogen-filled glove box. Diethyl ether and tetrahydrofuran were refluxed over sodium and benzophenone. Toluene was refluxed over sodium. Dichloromethane and acetonitrile were refluxed over calcium hydride. All solvents were distilled and degassed prior to use. IR spectra (nujol mulls, KBr or NaCl windows) were recorded on a Nicolet Avatar 360 FT IR spectrometer; 1H NMR spectra were recorded at room temperature on a Varian VXR 400 S spectrometer at 400 MHz or a Gemini 300 NMR spectrometer or a Bruker Advance DPX-300 spectrometer at 300 MHz. The 1H NMR spectra were calibrated against the residual protio impurity of the deuterated solvent. Elemental analyses were performed by the elemental analysis service at the London Metropolitan University. The ligand L1H4, L2H4 and L3H2 were prepared as described in the literature.5,23 The precursors [V(Np-RC6H4)(OtBu)3] (R = Me, CF3) were prepared via KtOBu using the method of Maatta.16 For the polymerization studies, the dry toluene employed as a polymerization solvent was purified by passage through columns of activated alumina and BASF R3-11 oxygen scavenger. Methylaluminoxane (MAO) was purchased from Albemarle Corporation as a 1.2 M toluene solution. This solution was dried under vacuum to remove the toluene and a substantial fraction of the AlMe3, to produce “dried MAO” (DMAO). Ethylene was obtained from Sumitomo Seika Co.Synthesis of {[VO(OnPr)(THF)]2(μ-p-L1)}·2(THF) (1·2(THF))
α,α,α′,α′-Tetrakis(3,5-di-tert-butyl-2-hydroxyphenyl)-p-xylene L1H4 (4.1 g, 4.4 mmol) was dissolved in tetrahydrofuran (40 mL). Vanadium oxytri-n-propoxide (1.0 mL, 4.5 mmol) was added via syringe and the solution was stirred at room temperature for 16 h. The volatiles were removed in vacuo, and crystallization using THF/light petroleum gave orange plates of the compound 1 (2.6 g, 45%). MS (EI, m/z) 1170.6 [M-2THF]+, 1110.5 [M-OnPr-2THF]+, 1068.5 [M-nPr-OnPr-2THF]2+. IR (Nujol, KBr, cm−1): 1597w, 1507m, 1435s, 1402w, 1286w, 1261m, 1221s, 1203s, 1153m, 1118s, 1104s, 1027s, 989s, 908m, 891m, 876s, 837s, 801m, 777m, 767m, 750m, 736w, 702w, 659s, 603m, 578w. Found: C, 71.61; H, 8.42. C70H100O8V2 (sample dried in vacuo for 24 h leads to loss of THF ×2) requires C, 71.77; H, 8.60%. 1H NMR (CDCl3): δ = 7.31 (d, 4H, J = 2.33, arylH), 7.22 (d, 4H, J = 2.33, arylH), 6.78 (s, 4H, arylH), 6.31 (s, 2H, Ar3-CH), 5.36 (t, 4H, J = 6.00, OCH2CH2), 3.75 (bm, 8H, THF α-H), 1.98 (sextet, 4H, J = 6.73, CH2CH2CH3), 1.86 (bm, 8H, THF β-H), 1.40 (s, 36H, tBu), 1.24 (s, 36H, tBu), 1.08 (t, 6H, J = 7.35, CH2CH3). 51V NMR (CDCl3): δ = −433.3 (w1/2 = 170 Hz).Synthesis of {L1[VO(tBuO)]2}·2MeCN (2·2MeCN)
L1H4 (4.1 g, 4.4 mmol) and [VO(tBuO)3] (3.20 g, 8.90 mmol) were refluxed in toluene (30 mL) for 12 h. On cooling, volatiles were removed in-vacuo and the residue can be extracted into either acetonitrile or dichloromethane (30 mL). Prolonged standing at 0 °C afforded 2 as a brown solid in 66% (3.72 g) yield. C72H104V2O8·0.75CH2Cl2 (sample dried in-vacuo for 2 h) requires C 69.17, H, 8.42. Found C, 68.84, H 8.77%. MS (solid, APCI):24 m/z 1199.6 [M]+, 1125.6 [M-OtBu]+, 1069.5 [M-OtBu-tBu]2+. IR: 1594w, 1568w, 1508w, 1401m, 1291m, 1261s, 1236m, 1224s, 1211m, 1200m, 1153s, 1118s, 1104s, 1021m, 1004s, 969bs, 911m, 876s, 845s, 826w, 800m, 777w, 768s, 731m, 722m, 705w, 668m, 653w, 644w, 604m, 542w, 497w, 467w. 1H NMR (CDCl3): δ = 7.30–7.21 (3× m, 8H, arylH), 6.75 (s, 4H, arylH), 6.34 (s, 2H, CH), 5.64 (s, 4H, 2× CH2Cl2), 1.69 (s, 18H, OC(CH3)3), 1.43 (s, 36H, C(CH3)3), 1.22 (s, 36H, C(CH3)3). 51V NMR (CDCl3) δ = −467.9 (w1/2 = 528 Hz). Single crystals of 2·2MeCN were grown from a saturated acetonitrile solution on prolonged standing at 0 °C.Synthesis of {L2[VO(OnPr)(THF)]2} (3)
As for 1, but using α,α,α′,α′-tetrakis(3,5-di-tert-butyl-2-hydroxyphenyl)-m-xylene (L2H4, 4.1 g, 4.4 mmol) and vanadium oxytri-n-propoxide (1.0 mL, 4.5 mmol). Crystallization using THF/light petroleum afforded orange needles of 3 (2.0 g, 35%). MS (E.I.): 1170.6 [M-2THF]+, 1110.5 [M-HOnPr-2THF]+. IR (Nujol, KBr, cm−1): 1595m, 1406m, 1361s, 1217s, 1154m, 1118s, 1104s, 1030s, 992s, 910w, 882w, 846s, 787s, 720s, 695w, 649s, 601m, 499w, 449w. Found: C, 71.60; H, 8.41. C70H100O8V2 (sample dried in vacuo for 24 h leads to loss of THF ×2) requires C, 71.76; H, 8.60%. 1H NMR (CDCl3): δ = 7.18 (d, 4H, J = 2.35, arylH), 7.15 (d, 4H, J = 2.35, arylH), 7.10 (s, 1H, arylH), 7.02 (t, 1H, J = 7.83, arylH), 6.80 (d, 2H, J = 7.93, arylH), 6.25 (s, 2H, Ar3-CH), 5.34 (t, 4H, J = 6.53, OCH2CH2), 3.74 (bm, 8H, THF α-H), 1.98 (sextet, 4H, J = 7.00, CH2CH2CH3), 1.86 (bm, 8H, THF β-H), 1.42 (s, 36H, tBu), 1.17 (s, 36H, tBu), 1.09 (t, 6H, J = 7.35, CH2CH3). 51V NMR (CDCl3) δ = −432.5 (w1/2 = 170 Hz).Synthesis of {[VO(tBuO)][V(Np-MeC6H4)(tBuO)](μ-p-L1)}·CH2Cl2 (4·CH2Cl2)
[V(Np-MeC6H4(OtBu)3] (3.3 g, 8.8 mmol) and L1H4 (4.1 g, 4.4 mmol) were refluxed in toluene (30 mL) for 12 h. On cooling, the volatiles were removed in-vacuo, and the residue was extracted into either acetonitrile (30 mL) or dichloromethane (30 mL). Cooling to −20 °C afforded 4 as small yellow/orange crystals. Yield 2.05 g, 34%. C79H111NO7V2·2CH2Cl2 requires C 66.70, H 7.95, N, 0.96%. Found C 66.22, H 8.32, N, 1.02%. MS (solvated with CH2Cl2 and diluted with MeCN for positive nano-electrospray technique): m/z 1291 [M]+, 1185 [M-H2Np–C6H4]+. IR: 1593w, 1568w, 1508w, 1403m, 1362s, 1291m, 1261s, 1238w, 1224m, 1201w, 1153m, 1115m, 1104s, 1018s, 974s, 912w, 876m, 844m, 800s, 778m, 769m, 737m, 705w, 669w, 644w, 603w, 573w, 542w, 468w. 1H NMR (CDCl3): δ = 7.22–7.13 (3× m, 12H, arylH), 6.68 (s, 4H, arylH), 6.27 (s, 2H, CH), 5.22 (s, 2H, CH2Cl2), 2.23 (s, 3H, tolylCH3), 1.93 (s, 3H, CH3CN), 1.69 (s, 18H, OC(CH3)3), 1.44 (overlapping s, 27H, C(CH3)3), 1.36 (s, 9H, C(CH3)3), 1.23 (overlapping s, 27H, C(CH3)3), 1.15 (s, 9H, C(CH3)3); 51V NMR (CDCl3) δ = −468.2 (w1/2 = 277 Hz), −558.5 (w1/2 = 297 Hz).Synthesis of {[VO(tBuO)][V(Np-CF3C6H4)(tBuO)](μ-p-L1)}·CH2Cl2 (5·CH2Cl2)
As for 7, but using [V(Np-CF3C6H4OtBu)3] (3.8 g, 8.8 mmol) and L1H4 (4.1 g, 4.4 mmol) affording on cooling (−20 °C) 5 as pale yellow/orange needles (yield 2.57 g, 41%). C79H106F3NO7V2·CH2Cl2 requires C, 67.40, H, 7.64, N, 0.98%. Found C 67.33, H 7.77, N 0.89%. MS (solvated with CH2Cl2 and diluted with MeCN for positive nano-electrospray technique): m/z 1342 [MH]+, 1269 [MH-OtBu]. IR: 1623w, 1599w, 1526w, 1507w, 1402m, 1375s, 1362s, 1327s, 1320s, 1292m, 1259s, 1237m, 1213m, 1200m, 1152s, 1117s, 1104s, 1066s, 976s, 911m, 873m, 841s, 801s, 768s, 755w, 735w, 720w, 705w, 696w, 667m, 652w, 643w, 621w, 597m, 573w, 540w, 503w, 494w. 1H NMR (CDCl3): δ = 7.33–7.13 (3× m, 8H, arylH), 6.68 (s, 4H, arylH), 6.27 (s, 2H, CH), 5.22 (s, 2H, CH2Cl2), 1.93 (s, 3H, CH3CN), 1.69 (s, 18H, OC(CH3)3), 1.53 (s, 9H, C(CH3)3), 1.37 (s, 27H, C(CH3)3), 1.23 (s, 9H, C(CH3)3), 1.17 (s, 27H, C(CH3)3); 19F NMR (CDCl3) δ = −61.2; 51V NMR (CDCl3) δ = −467.7 (w1/2 = 520 Hz), −539.6 (w1/2 = 296 Hz).Synthesis of {[V(THF)(Np-MeC6H4)Cl]2(μ-p-L1)} (6·4toluene)
L1H4 (1.00 g, 1.08 mmol) and Na (0.10 g, 4.35 mmol) were stirred in THF (30 mL) at ambient temperature for 12 h. The solution was then cooled to −78 °C and solid [V(Np-MeC6H4)Cl3] (0.60 g, 2.29 mmol) was added. The mixture was allowed to warm to room temperature and was stirred for 12 h. Volatiles were removed in-vacuo, and the residue was extracted into toluene (30 mL) or dichloromethane (30 mL). Brown/red prisms of 6 were formed on prolonged standing (1–2 days) at 0 °C. Yield: 1.61 g, 83%. C87H102Cl2N2O6V2 (sample crystallized from CH2Cl2, 6·CH2Cl2) requires C, 68.95, H, 6.78, N, 1.84%. Found C 69.92*, H 7.16, N 2.05%. *Despite repeated attempts, we were unable to obtain closer %C values. MS (EI, positive mode): m/z 1527 (M+–Cl–THF), 1459 (M+–2Cl–tolylNH2). IR: 3544w, 1597w, 1568m, 1506w, 1291m, 1261s, 1240m, 1224m, 1201w, 1153w, 1118m, 1104m, 1007s, 968s, 912w, 876m, 844m, 799m, 778w, 768m, 722s, 668w, 643w, 603w, 496w, 462w. 1H NMR (CDCl3): δ = 7.45–6.92 (3× m, 17H, arylH including one toluene), 6.56 (s, 2H, CH), 6.49 (d, J 8.0 Hz, 4H, arylH), 5.97 (d, J 8.0 Hz, 4H, arylH), 4.29 (m, 8H, THF α-H), 2.37 (s, 6H. CH3C6H4), 2.17 (s, 3H, CH3 of toluene), 2.02 (m, 8H, THF β-H), 1.31 (s, 36H, C(CH3)3), 1.23 (s, 36H, C(CH3)3); 51V NMR (CDCl3) δ = −7.3 (w1/2 = 3373 Hz).Synthesis of {[V(THF)(Np-CF3C6H4)Cl]2(μ-p-L1)} 7
As for 8, but using [V(Np-CF3C6H4)Cl3] (0.72 g, 2.28 mmol) and L1H4 (1.00 g, 1.08 mmol) and Et3N (0.63 mL, 4.55 mmol) affording red/brown 7; the complex can be recrystallized from cold toluene or dichloromethane, yield 0.71 g, 47%. C114H126Cl2F6N2O6V2·CH2Cl2 requires C, 63.73, H, 6.89, N, 1.71%. Found C 63.56, H 7.20, N 1.47%. MS (EI, positive mode) 1411.5 [MH–2THF]+, 1340.6 [MH–2THF–2Cl]+, 1250 [MH–2THF–H2NC6H4CF3]+, 1089 [MH–2THF–2H2NC6H4CF3]+. IR: 2335w, 1693w, 1651w, 1616w, 1600w, 1507w, 1406m, 1364s, 1321s, 1261s, 1240w, 1226m, 1201w, 1168s, 1122s, 1104s, 1066s, 1021s, 956w, 910w, 873m, 841s, 803s, 773m, 753w, 728s, 692m, 668w, 645w, 626w, 596m, 556w, 502w, 464w. 1H NMR (CDCl3): δ = 7.54–7.02 (3× m, 16H, arylH + imidoarylH), 6.76 (s, 4H, arylH), 6.35 (s, 2H, CH), 5.29 (s, 2H, CH2Cl2), 3.12 (bm, 8H, THF α-H), 1.85 (bm, 8H, THF β-H), 1.43 (s, 36H, C(CH3)3), 1.23 (s, 36H, C(CH3)3); 19F NMR (CDCl3) δ = −60.7; 51V NMR (C6D6) δ = −211.1 (w1/2 = 647 Hz).Synthesis of {L3(VO)2(μ-OnPr)2} (8)
2-(α-(2-Hydroxy-3,5-di-tert-butylphenyl)benzyl)-4,6-di-tert-butylphenol (L3H2, 4.1 g, 8.2 mmol) was dissolved in tetrahydrofuran (40 mL). Vanadium oxytri-n-propoxide (1.9 mL, 8.4 mmol) was added via syringe and the solution was stirred at room temperature for 16 h. The volatiles were removed in vacuo, following which crystallization using warm light petroleum gave red needles of the vanadium dimer 8 (3.6 g, 70%). MS (EI, m/z): 624.4 [M]+, 564.4 [M-OnPr]. IR (Nujol, KBr, cm−1): 1599m, 1381m, 1289w, 1220s, 1153s, 1103s, 1060s, 989s, 911w, 875w, 855m, 836s, 800m, 770m, 745m, 705m, 652s, 599m, 503w, 451w. Found: C, 72.82; H, 8.39. C76H106O8V2 requires C, 73.05; H, 8.55%. 1H NMR (CDCl3): δ = 7.33 (d, 4H, J = 2.30, arylH), 7.27 d, (4H, J = 2.30, arylH), 7.21–7.15 (m, 4H, arylH), 7.00 (d, 4H, J = 7.95, arylH), 6.38 (s, 2H, Ar3CH), 5.37 (t, 4H, J = 6.03, OCH2CH2), 1.99 (sextet, 4H, CH2CH2CH3), 1.47 (s, 36H, tBu), 1.26 (s, 36H, tBu), 1.10 (t, 6H, J = 7.40, CH2CH3). 51V NMR (CDCl3): δ = −433.6 (w1/2 = 170 Hz).Polymer characterization
The melt transition temperatures (Tm) of the polyethylene (PE) and ethylene/propylene copolymer (EPR) were determined by differential scanning calorimetry (DSC) with a Shimadzu DSC-60 instrument. The polymer samples were heated at 50 °C min−1 from 20 °C to 200 °C, held at 200 °C for 5 min, and cooled to 0 °C at 20 °C min−1. The samples were held at this temperature for 5 min, and then reheated to 200 °C at 10 °C min−1. The reported Tm was determined from the second heating scan unless otherwise noted.Molecular weights (Mw and Mn) and molecular weight distributions (MWDs) of PE and EPR were determined using a Waters GPC2000 gel permeation chromatograph equipped with four TSKgel columns (two sets of TSKgelGMH6-HT and two sets of TSKgelGMH6-HTL) at 140 °C using polyethylene calibration. o-Dichlorobenzene (ODCB) was used as the solvent.
Polymerization procedure
Polymerization reactions were performed in a parallel pressure reactor (Argonaut Endeavor® Catalyst Screening System) containing 8 reaction vessels (15 mL) each equipped with a mechanical stirrer and monomer feed lines. At first, a toluene solution (and a toluene solution of ETA where necessary) were injected into each vessel. For ethylene polymerization, the solution was heated to the polymerization temperature (Tp) and thermally equilibrated, and the nitrogen atmosphere was replaced with ethylene and the solution was saturated with ethylene at the polymerization pressure. For ethylene/propylene copolymerization, the solution was heated to the Tp and thermally equilibrated, and the nitrogen atmosphere was replaced with propylene and the reaction vessels were pressurized with propylene (0.4 MPa at 25 °C), then ethylene was introduced into the reactor up to the polymerization pressure. In all cases the polymerization was started by addition of a toluene solution of alkyl aluminum or alkyl aluminum chloride followed by addition of a toluene solution of the vanadium complex (0.50 mL toluene solution of complex followed by 0.25 mL toluene wash). The total volume of the reaction mixture was 5 mL for all polymerizations. The pressure was kept constant by feeding ethylene on demand. After the reaction, the polymerization was stopped by addition of excess isobutyl alcohol. The resulting mixture was added to acidified methanol (45 mL containing 0.5 mL of concentrated HCl). The polymer was recovered by filtration, washed with methanol (2 × 10 mL) and dried in a vacuum oven at 80 °C for 10 h.Crystallography
Single crystal X-ray diffraction data for 7·toluene·THF were collected in series of ω-scans using a Stoe IPSD2 image plate diffractometer utilising monochromated Mo radiation (λ = 0.71073 Å). Standard procedures were employed for the integration and processing of the data using X-RED.25 Samples were coated in a thin film of perfluoropolyether oil and mounted at the tip of a glass fibre located on a goniometer. Data were collected from crystals held at 150 K in an Oxford Cryosystems nitrogen gas cryostream.All other single crystal data were collected using series of ω-scans by the EPSRC UK National Crystallography Service; data for 3 was collected at the Diamond Light Source (I19, synchrotron radiation λ = 0.6889 Å); data for the remaining samples were collected using a radiation from a Mo rotating anode source. Samples were mounted using MiTeGen loops and held at 100 K in an Oxford Cryosystems nitrogen gas cryostream. Data were corrected for Lp effects and for absorption. CrystalClear-SM Expert 3.1 b27 (Rigaku, 2012); cell refinement: CrystalClear-SM Expert 3.1 b27 (Rigaku, 2012); data reduction: CrystalClear-SM Expert 3.1 b27 (Rigaku, 2012); program(s) used to solve structure: SHELXS97;26 program(s) used to refine structure: SHELXL2013.26
Many of the structures contained small-scale disorder involving the t-butyl groups. This was handled using standard procedures. The SQUEEZE27 routine was applied to model scattering from regions of disordered solvent in these structures: 3 and 8. The crystal examined for 6·MeCN was found to be twinned and this was handled by routine techniques and the final model refined using the HKLF5 formalism using all observed data.
Acknowledgements
The EPSRC Mass Spectrometry Service (Swansea, UK) and the EPSRC National X-ray Crystallographic Service (Southampton) are thanked for data collection. CR thanks the EPSRC for an Overseas Travel grant.References
- (a) R. Galletti and G. Pampaloni, Coord. Chem. Rev., 2010, 254, 525–536 CrossRef PubMed; (b) K. Nomura and W. Zhang, Chem. Sci., 2010, 1, 161–173 RSC; (c) Y. R. Patil, in Olefins Polymerization Reactivity of Niobium-Based Metal Complexes, Lambert Academic Publishing, US, 2011 Search PubMed; (d) D. Wang, Z. Zhao, T. B. Mikenas, X. Lang, L. G. Echevskaya, C. Zhao, M. A. Matsko and W. Wu, Polym. Chem., 2012, 3, 2377–2382 RSC.
- See for example: (a) H. Hagen, J. Boersma and G. van Koten, Chem. Soc. Rev., 2002, 31, 357–364 RSC; (b) S. Gambarotta, Coord. Chem. Rev., 2003, 237, 229–243 CrossRef CAS; (c) Y. Onishi, S. Katao, M. Fujiki and K. Nomura, Organometallics, 2008, 27, 2590–2596 CrossRef CAS; (d) J. Q. Wu, L. Pan, N. H. Hu and Y. S. Li, Organometallics, 2008, 27, 3840–3848 CrossRef CAS; (e) C. Redshaw, Dalton Trans., 2010, 39, 5595–5604 RSC and references therein.
- (a) D. Takeuchi, Dalton Trans., 2010, 39, 311–328 RSC; (b) K. Nomura and W. Zhang, Chem. Rev., 2011, 111, 2342–2362 CrossRef CAS PubMed; (c) J.-Q. Wu and Y.-S. Li, Coord. Chem. Rev., 2011, 255, 2303–2314 CrossRef CAS PubMed; (d) S. W. Zhang, L.-P. Lu, B.-X. Li and Y.-S. Li, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4721–4731 CrossRef CAS PubMed; (e) S. W. Zhang, L.-P. Lu, Y.-Y. Long and Y.-S. Li, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 844–854 CrossRef CAS PubMed; (f) L.-P. Lu, J.-B. Wang, J.-Y. Liu and Y.-S. Li, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2633–2642 CrossRef CAS PubMed.
- (a) C. Redshaw, L. Warford, S. H. Dale and M. R. J. Elsegood, Chem. Commun., 2004, 1954–1956 RSC; (b) C. Redshaw, M. A. Rowan, D. M. Homden, S. H. Dale, M. R. J. Elsegood, S. Matsui and S. Matsuura, Chem. Commun., 2006, 3329–3331 RSC; (c) C. Redshaw, M. A. Rowan, L. Warford, D. M. Homden, A. Arbaoui, M. R. J. Elsegood, S. H. Dale, T. Yamato, C. P. Casas and S. Matsui, Chem.–Eur. J., 2007, 13, 1090–1107 CrossRef CAS PubMed; (d) D. M. Homden, C. Redshaw, L. Warford, D. L. Hughes, J. A. Wright, S. H. Dale and M. R. J. Elsegood, Dalton Trans., 2009, 8900–8910 RSC; (e) C. Redshaw, D. M. Homden, D. L. Hughes, J. A. Wright and M. R. J. Elsegood, Dalton Trans., 2009, 1231–1242 RSC; (f) L. Clowes, C. Redshaw and D. L. Hughes, Inorg. Chem., 2011, 50, 7838–7845 CrossRef CAS PubMed; (g) C. Redshaw, L. Clowes, D. L. Hughes, M. R. J. Elsegood and T. Yamato, Organometallics, 2011, 30, 5620–5624 CrossRef CAS; (h) C. Redshaw, M. J. Walton, D. S. Lee, C. Jiang, M. R. J. Elsegood and K. Michiue, Chem.–Eur. J., 2015, 21, 5199–5210 CrossRef CAS PubMed; (i) C. Redshaw, M. J. Walton, K. Michiue, Y. Chao, A. Walton, P. Elo, V. Sumerin, C. Jiang and M. R. J. Elsegood, Dalton Trans., 2015, 44, 12292–12303 RSC.
- L. H. Tang, E. P. Wasserman, D. R. Neithamer, R. D. Krystosek, Y. Cheng, P. C. Price, Y. Y. He and T. J. Emge, Macromolecules, 2008, 41, 7306–7315 CrossRef CAS.
- J. Zhang, C. Jian, Y. Gao, L. Wang, N. Tang and J. Wu, Inorg. Chem., 2012, 51, 13380–13389 CrossRef CAS PubMed.
- Y. Al-Khafaji, X. Sun, T. J. Prior, M. R. J. Elsegood and C. Redshaw, Dalton Trans., 2015, 44, 12349–12356 RSC.
- A. G. Fisch, J. N. da Silveira, N. S. M. Cardozo, A. R. Secchi, J. H. Z. dos Santos and J. B. P. Soares, J. Mol. Catal. A: Chem., 2013, 366, 74–83 CrossRef CAS PubMed.
- R. E. LaPointe, J. C. Stevens, P. N. Nickias and M. H. McAdon, Dow Chemicals US., EP 0 520 732 B1, 1992.
- (a) R. Figueroa, R. D. Froese, Y. He, J. Klosin, C. N. Theriault and K. A. Abboud, Organometallics, 2011, 30, 1695–1709 CrossRef CAS; (b) P. P. Fontaine, R. Figueroa, S. D. McCann, D. Mort and J. Klosin, Organometallics, 2013, 32, 2963–2972 CrossRef CAS; (c) T. R. Boussie, G. M. Diamond, C. Goh, K. A. Hall, A. M. LaPointe, M. K. Leclerc, V. Murphy, J. A. W. Shoemaker, H. Turner, R. K. Rosen, J. C. Stevens, F. Alfano, V. Busico, R. Cipullo and G. Talarico, Angew. Chem., Int. Ed., 2006, 45, 3278–3283 CrossRef CAS PubMed; (d) P. S. Chum and K. W. Swogger, Prog. Polym. Sci., 2008, 33, 797–819 CrossRef CAS PubMed; (e) For a recent review on the use of Post-Metallocenes catalysts for industrial polyolefin production, see:M. C. Baier, M. A. Zuideveld and S. Mecking, Angew. Chem., Int. Ed., 2014, 53, 9722–9744 CrossRef CAS PubMed.
- (a) N. Desmangles, S. Gambarotta, C. Bensimon, S. Davis and H. Zahalka, J. Organomet. Chem., 1998, 562, 53–60 CrossRef CAS; (b) Y. Ma, D. Reardon, S. Gambarotta and G. Yap, Organometallics, 1999, 18, 2773 CrossRef CAS; (c) H. Hagen, J. Boersma, M. Lutz, A. L. Spek and G. van Koten, Eur. J. Inorg. Chem., 2001, 117 CrossRef CAS; (d) M. Arndt-Rosenau, M. Hoch, J. Sundermeyer, J. Kipke and X. Li, US Pat., 01 304551, 2003; (e) Y. Nakayama, H. Bando, Y. Sonobe and T. Fujita, J. Mol. Catal. A: Chem., 2004, 213, 141 CrossRef CAS PubMed; (f) S. Cuomo, S. Milione and A. Grassi, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3279 CrossRef PubMed; (g) S. Zhang, W.-C. Zhang, D.-D. Shang, Z.-Q. Zhang and Y.-X. Wu, Dalton Trans., 2015, 44, 15264–15270 RSC.
- See for example: (a) C. R. Randall and W. H. Armstrong, J. Chem. Soc., Chem. Commun., 1988, 986–987 RSC; (b) G. Asgedom, A. Sreedhara, J. Kivikoski, E. Kolehmainen and C. P. Rao, J. Chem. Soc., Dalton Trans., 1996, 93–97 RSC.
- (a) M. Mazzanti, C. Floriani, A. Chiese-Villa and C. Guastini, J. Chem. Soc., Dalton Trans., 1989, 1793–1798 RSC; (b) P. J. Toscano, E. J. Schermerhorn, C. Dettelbacher, D. Macherone and J. Zubieta, J. Chem. Soc., Chem. Commun., 1991, 933–934 RSC.
- (a) W. Clegg, M. R. J. Elsegood, S. J. Teat, C. Redshaw and V. C. Gibson, J. Chem. Soc., Dalton Trans., 1998, 3037–3040 RSC; (b) W. Clegg, J. Chem. Soc., Dalton Trans., 2000, 3223–3232 RSC.
- (a) C. Redshaw, in Olefin Upgrading Catalysis by Nitrogen-based Metal Complexes I, ed. G. Giambastini and J. Cámpora, Springer, 2011 Search PubMed; (b) S. Zhang and K. Nomura, Catal. Surv. Asia, 2011, 15, 127–133 CrossRef CAS.
- (a) E. A. Maatta, Inorg. Chem., 1984, 23, 2561–2562 CrossRef; (b) D. D. Devore, J. D. Lichtenhan, F. Takusagawa and E. A. Maatta, J. Am. Chem. Soc., 1987, 109, 7408–7416 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef PubMed.
- Spiro compounds related to motif I in the CSD: (a) A. M. Litwak, F. Grynazpan, O. Aleksiuk, S. Cohen and S. E. Biali, J. Org. Chem., 1983, 58, 393–402 CrossRef; (b) F. Grynszpan and S. E. Biali, J. Chem. Soc., Chem. Commun., 1994, 2545–2546 RSC; (c) O. Aleksiuk, S. Cohen and S. E. Biali, J. Am. Chem. Soc., 1995, 117, 9645–9652 CrossRef CAS; (d) F. Grynszpan and S. E. Biali, J. Org. Chem., 1996, 61, 9512–9521 CrossRef CAS; (e) J. Wöhnert, J. Brenn, M. Stoldt, O. Aleksiuk, F. Grynszpan, I. Thondorf and S. E. Biali, J. Org. Chem., 1998, 63, 3866–3874 CrossRef; (f) K. Agharia, O. Aleksiuk, S. E. Biali, V. Böhmer, M. Frings and I. Thondorf, J. Org. Chem., 2001, 66, 2891–2899 CrossRef PubMed; (g) K. Agharia and S. E. Biali, J. Am. Chem. Soc., 2001, 123, 12495–12503 CrossRef PubMed.
- A. G. Maestri and S. N. Brown, Inorg. Chem., 2004, 43, 6996–7004 CrossRef PubMed.
- For early use of ETA, see: (a) A. Gumboldt, J. Helberg and G. Schleitzer, Makromol. Chem., 1967, 101, 229–245 CrossRef CAS PubMed; (b) D. L. Christman, J. Polym. Sci., Part A: Polym. Chem., 1972, 10, 471–487 CrossRef CAS PubMed; (c) E. Addison, A. Deffieux, M. Fontanille and K. Bujadoux, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 1033–1041 CrossRef PubMed.
- E. W. Hansen, R. Blom and O. M. Bade, Polymer, 1997, 38, 4295–4304 CrossRef CAS.
- W. Wang and K. Nomura, Macromolecules, 2005, 38, 5905–5913 CrossRef CAS.
- E. Müller, A. Schick and R. Mayer, Chem. Ber., 1960, 93, 2649–2662 CrossRef PubMed.
- APCI refers to the ionization method, Atmospheric Pressure Chemical Ionization, in which samples introduced into the APCI source via an Atmospheric Solids Analysis Probe (ASAP) are vaporized then ionized using a corona discharge (∼4μA). The benefit of using ASAP is that samples can be introduced at ambient temperature as solids or in solution, then the temperature in the source is increased until the sample vaporizes.
- X-AREA v. 1.64, STOE & Cie GmbH, Darmstadt, 2012 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, C34 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic files CIF format for the structure determinations of compounds 1·2THF, 2·2MeCN, 3, 4·CH2Cl2, 5·CH2Cl2, 6·4toluene, 7 and 8. CCDC 1404522-3, 1404527-8, 1404530-2. CCDC 1404522-3, 1404527-8, 1404530-2 (structures 1 & 3, 4 & 5, 6–8) and 1048777 (structure 2) contain the supplementary crystallographic, 51V spectroscopic and polymerization data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra20177b |
| This journal is © The Royal Society of Chemistry 2015 |