
Thermal-based regulation on biomineralization and biological properties of bioglass nanoparticles decorated PAN-based carbon nanofibers
Abstract
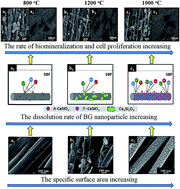
Composite carbon nanofibers (CNFs) containing bioglass (BG) nanoparticles displayed different morphology and microstructures depending on the sintering temperature (800, 1000 and 1200 °C) when they were produced from an electrospun polyacrylonitrile–BG precursor blend nanofibers. Biomineralization using simulated body fluid (SBF) and biological evaluation using an osteoblast culture were performed to investigate their relationship with sintering temperature. Characterization revealed that the BG nanoparticles on CNF/BG sintered at 1000 °C (CNF/BG-1000) possessed small particle size and uniform size distribution, and the crystallinity of the BG nanoparticles increased as the sintering temperature was increased from 800 to 1200 °C. The dissolution rate of the BG nanoparticles was thus different between the cases, which enhanced the biomineralization and cell proliferation/differentiation to varying degrees. Benefiting from the homogeneous distribution and large specific surface area of the BG nanoparticles on the CNFs, the results demonstrated that CNF/BG-1000 had the strongest ability in promoting apatite deposition, proliferation and osteogenic differentiation of MC3T3-E1 pre-osteoblasts in comparison with CNF/BG sintered at 800 or 1200 °C. The results demonstrate a flexible tool to regulate the physiochemical and biological properties of CNF/BG composites by controlling the sintering temperature, which could find promising applications in skeleton repairing.
 Please wait while we load your content...
Please wait while we load your content...

