
Controlled nitridation of tantalum (oxy)nitride nanoparticles towards optimized metal-support interactions with gold nanocatalysts†
Xiaoyun Yanga,
Sina Hea,
Yijin Shua,
Zhangping Shib,
Yulin Guoa,
Qingsheng Gao*a and
Yi Tangb
aDepartment of Chemistry, Jinan University, 510632, Guangzhou, China. E-mail: tqsgao@jnu.edu.cn
bDepartment of Chemistry, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Laboratory of Advanced Materials and Collaborative Innovation Center of Chemistry for Energy Materials, Fudan University, 200433, Shanghai, China
First published on 14th October 2015
Abstract
The electron regulation on supports can vary metal-support interactions with loaded metals in heterogeneous catalysis. In this paper, a facile Sr2+-mediated ionothermal route was introduced to control the nitridation degree in tantalum (oxy)nitrides, resulting in varied electronic properties and optimized interactions with gold nanocatalysts. A new mechanism was proposed that the formation of SrTa4O11 intermediates facilitated the replacement of O by N in controlled nitridation, and more importantly avoided undesired over-nitridation. As expected, the TaON support with defined nitridation promoted electronic metal-support interactions to generate Auδ− species, which was highly active for the thermal hydrogenation of nitrobenzene due to the moderated adsorption and effective activation on Auδ− in Au/TaON. This work elucidated the optimized metal-support interactions achieved on controllably nitridated supports, opening up new opportunities for the development of efficient nanocatalysts.
1. Introduction
Metal nitrides are a class of important functional materials in catalysis, refractory ceramics, sensing, electrochemistry, and optoelectronics, due to their rich physicochemical properties associated with the tunable interactions between metal and nitrogen.1–4 On one hand, the enlarged metal–metal bonds in the alloy phases of many metal nitrides lead to the reduced density of unoccupied d-orbitals around Fermi level, and the noble-metal-like catalytic behaviours.5–8 On the other hand, the electron transfer from metal to nitrogen will oppositely increase the density of unoccupied d-orbitals.8 Thus, the synergy of the above two functions varies the electronic features and catalytic performance of metal nitrides. Many nitrides active for hydrogen-involving catalytic reactions have been developed,5,7 and very recently, they are discovered as prominent supports due to the rich interactions with loaded metals.9,10 As evidence in catalytic nitroarene hydrogenation,11,12 the metal-support interactions with gold are promoted by Mo2N supports. In comparison with the counterparts of metal oxides,13,14 several advantages are highlighted in nitride-based supports, e.g., noble-metal-like d-band structures, tailorable electron properties, and tunable metal-support interactions.7 Regarding the prevailing Mott–Schottky effects in the interfaces between metals and semiconductors,15 the electron regulation on nitrides is desired to further optimize the metal-support interactions, which is however rarely reported.Introducing N into metal oxides usually brings narrowed band-gap and increased Fermi level as a result of the synergetic effects of both alloying and electron-transfer,16,17 which indicates a facile electron-manipulation by controlled nitridation. In the case of tantalum (oxy)nitrides, the smaller band-gap energy (vs. Ta2O5) enables them as novel visible-light photocatalysts,17,18 and the rich defect and tunable valence state are further proved promising for nonphotocatalytic hydrogenation and oxidation.19–21 It's more important that tantalum (oxy)nitrides can be utilized as promising catalyst supports. Their increased Fermi level and tunable electron features would promote the electronic interactions with metals, showing advantages in comparison with Ta-oxides.22–24 To achieve the optimized metal-support interactions in efficient catalysis, the controlled nitridation in tantalum (oxy)nitrides is highly demanded, however, it suffers easy over-nitridation to by-products of Ta3N5, Ta4N5, and even TaN during high-temperature synthesis.25,26
Nitridation utilizing organic–inorganic hybrid precursors provides the tailored generation of nitrides.8,27 Herein, a facile Sr2+-mediated route is conducted to accomplish the controlled nitridation in tantalum (oxy)nitride nanoparticles, which are mono-dispersed, well-separated and mostly single crystalline. A new mechanism via tantalate intermediate is proposed for the controlled nitridation. The formation of SrTa4O11, in which Ta–O bonds are weakened by Sr2+, ensures the generation of pure TaON or Ta3N5 at mild temperature, and avoids over-reactions. This innovation can be extended to other ionothermal system utilizing a wide range of alkaline-earth-metal salts, and can well address the remaining puzzlements in the previous work. For example, the previous Ca2+-assisted mechanism associated with the controlled NH3-release from Ca2+-chelated urea is controversial,28 because urea decomposes much earlier than nitridation reactions upon heating. The new mechanism via tantalate intermediates makes an obvious progress in the understanding of essential effects of ionic solvents. More importantly, it is discovered for the first time that such controlled nitridation obviously varies the electronic properties of tantalum (oxy)nitrides to express electronic metal-support interactions with gold for catalytic hydrogenation. In the probe reaction of nitrobenzene (NB) reduction under thermal condition, remarkably improved activity and optimized aniline (AN) selectivity are achieved over Au/TaON, as compared with those on Au/Ta2O5 and Au/Ta3N5. This is ascribed to the suitable adsorption and effective activation of substrates by moderately negative Auδ− on TaON support.
2. Experimental section
2.1 Catalysts preparation
In a typical ionothermal procedure, 0.250 g of TaCl5 powder was added to 2 mL of methanol, followed by adding 0.103 of SrCO3. After a varied amount of urea was added, the solution was let dry to gel under stirring at room temperature. The precursor was transferred into an oven and kept under N2 flow (500 mL min−1) for 2.0 h in order to remove air before heating, and then was heated to 700 °C with a ramping rate of 5 °C min−1, being held for 5 h. After treating the as-received powders with 1 M HCl (aq.) for 48 hours to remove Sr-species, tantalum (oxy)nitrides nanoparticles were obtained. Other ionothermal synthesis employing Ba2+, Ca2+ and Mg2+ was carried out via the same processes, except that SrCO3 was replaced by BaCO3 (0.138 g), CaCO3 (0.070 g) and MgCO3 (0.059 g), respectively.Meanwhile, Ta2O5 nanoparticles used for control catalytic tests were synthesized via traditional reverse homogeneous precipitation. 0.360 g of TaCl5 was dissolved in 5 mL of methanol, and then, 15 mL of NH3·H2O (19%, aq.) was added. After stirring for 1 hour, the white precipitate was washed with water and ethanol. After heating at 750 °C for 5 h under air, Ta2O5 nanoparticles were received.
A typical deposition–precipitation procedure was employed to prepare Au/Ta2O5, Au/TaON, and Au/Ta3N5 catalysts. Briefly, Ta-based nanoparticles were dispersed with the aqueous solution of HAuCl4 (4.856 × 10−3 mol L−1), and pH was adjusted to 9.0 by dropwise addition of 0.25 M NH3·H2O (aq.). After stirring for 6 hours and aging for another 2 hours, the catalyst was washed with deionized water for five times and then dried at 50 °C overnight, followed by a careful reduction with a stream of 5 vol% H2/Ar at 300 °C for 2 hours.
2.2 Physical measurement
X-ray diffraction (XRD) analysis was performed on Bruker D8 diffractometer using Cu Kα radiation (λ = 1.54056 Å). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) investigations were taken on a ZEISS ULTRA55 and a JEOL JEM 2100F, respectively. Energy dispersive spectrum (EDS) was carried out on a JEOL JEM 2100F. X-ray photoelectron spectroscopy (XPS) analysis was processed on a Perkin-Elmer PHI X-tool, using C 1s (B. E. = 284.6 eV) as a reference. The N content in TaON and Ta3N5 was determined by CHNS elemental analysis using a Vario EL Elementar, and the Au loading was investigated by an inductively coupled plasma-atomic emission spectroscopy (ICP-AES). Thermogravimetric analysis coupling with differential scanning calorimeter (TGA/DSC) was tested on NETZSCH STA449F3 under N2 flow. The UV-vis diffuse reflection spectra (UV-vis DRS) were carried out on Varian Cary 5000 at room temperature. IR spectra were collected with a Nicolet 6700 FTIR spectrometer. The B.E.T. specific surface areas were determined by adsorption–desorption of nitrogen at liquid nitrogen temperature, using a Micromeritics TriStar 3000 equipment, degassing at 300 °C. Hydrogen temperature-programmed reduction (H2-TPR) and CO pulse adsorption were both conducted on the XianQuan instrument TP 5076.2.3 Catalytic test of NB hydrogenation
Nonphotocatalytic test was carried out in a 100 mL stainless steel autoclave, in which 40 mg of catalyst, 0.5 mmol of NB, 6.0 mmol of formic acid (FA), 5.0 mL of H2O and 5.0 mL of EtOH were loaded. The reactor was purged with 0.5 MPa N2. The reaction was conducted at 80 °C for 2.5 hours, with the stirring of 500 rpm. The products were analyzed by Shimadzu HPLC LC-20A with a RID detector. To further study the catalytic mechanism, a series of control tests by photo-excitation were conducted in a stainless steel autoclave with a quartz window under the irradiation of Xe lamp (λ > 200 nm adopted for Au/Ta2O5, and λ = 400–800 nm for Au/TaON and Au/Ta3N5). Meanwhile, in the catalytic decomposition of FA over supported Au, the liquid product was analyzed by Shimadzu HPLC LC-20A, and the gas product was tested by Shimadzu GC-2014C.3. Results and discussion
3.1 Characterization of TaON and Ta3N5 nanoparticles
In the presence of SrCl2, the controllable nitridation of tantalum can be achieved via simply changing the molar ratio of urea to Ta (RU/Ta) in starting gels, followed by calcination at 700 °C under N2 flow and a treatment with HCl. As confirmed by XRD investigation (Fig. 1), the products evolve from Ta2O5 to TaON and finally Ta3N5 as RU/Ta is increased from 1.0 to 7.0, accompanied with the visible color changing from white to yellow and finally red. For a RU/Ta of 2.0, TaON products are obtained, namely γ-TaON (ICDD no. 01-076-3258) and β-TaON (ICDD no. 04-010-4352); while, Ta3N5 (ICDD no. 01-089-5200) is harvested as RU/Ta is 7.0, indicating the well-defined nitridation. The comparison of these products with the standard patterns from ICDD database is further shown in ESI as Fig. S1.† | ||
| Fig. 1 XRD patterns of products obtained in the presence of Sr2+ with the RU/Ta from 1.0 to 7.0. | ||
Furthermore, the SEM investigation shows that all the products are in form of well-defined nanoparticles (Fig. S2 in ESI†). Representatively, Fig. 2a and b display the SEM images of TaON and Ta3N5, which are obtained with a typical RU/Ta of 2.0 and 7.0, respectively. They are about 20–30 nm in diameter. In the TEM images (Fig. 2c and d), TaON can be distinguished by the clear (110) and (200) lattice fringes, while Ta3N5 is confirmed by the (110) and (402). The EDS obtained on the both of above nanoparticles confirm the complete removal of SrCl2 by a treatment with 1 M HCl (Fig. S3 in ESI†). Additionally, such TaON and Ta3N5 exhibit a surface area of 21.3 and 25.6 m2 g−1, respectively (Table S1 in ESI†).
 | ||
| Fig. 2 (a and b) SEM and (c and d) TEM images of (a and c) TaON and (b and d) Ta3N5 NPs, harvested at RU/Ta = 2.0 and 7.0, respectively. | ||
XPS analysis further gives clear evidence about TaON and Ta3N5 with varied electronic structures (Fig. 3). With the introduction of N, TaON presents the obviously shifted peaks of Ta 4f7/2 and 4f5/2 at the lower binding energies of 26.0 and 27.7 eV, respectively, as compared with those located at 26.9 and 28.7 eV in Ta2O5.29 This is associated with the lower electronegativity of N than that of O, as well as the consequently favored electron transfer from N to Ta. In this way, Ta3N5 displays the further decreased binding energy of 25.1 and 26.9 eV for Ta 4f7/2 and 4f5/2, respectively. Meanwhile, the red-shifted absorption bands of TaON and Ta3N5 nanoparticles in UV/vis DRS (Fig. S4a in ESI†), in comparison with those for Ta2O5, indicate the narrowed band-gap due to nitridation. According to previous reports,29 the valence band in tantalum (oxy)nitrides, mainly contributed by O 2p and N 2p orbitals, ascends from −7.93 eV in Ta2O5, to −6.6 eV in TaON and −6.02 eV in Ta3N5, bringing about obviously increased Fermi level with the increased nitridation degree (Fig. S4b in ESI†).
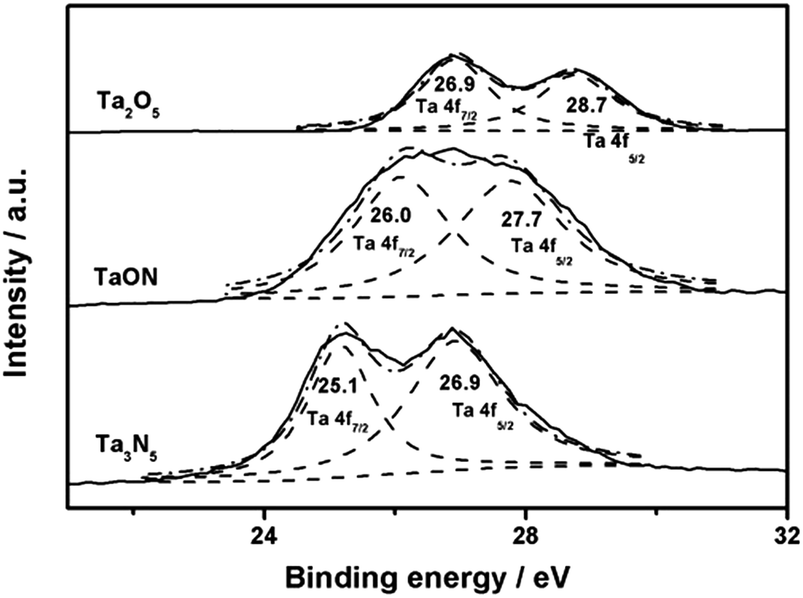 | ||
| Fig. 3 Ta 4f XPS spectra of Ta2O5, TaON and Ta3N5. | ||
3.2 Mechanism for the controlled nitridation of tantalum
Sr2+ is found indispensable for the effective control over nitridation. In the absence of Sr2+, uncontrolled nitridation usually results in undesired mixtures (Fig. 4). With a low RU/Ta of 1.0, un-nitridated Ta2O5 and over-nitridated Ta3N5 are observed along with desired TaON. As RU/Ta is increased from 2.0 to 7.0, the mixture of TaON and Ta3N5 unfortunately remains, and at a high RU/Ta of 8.0, another by-product of TaN emerges. Given the rich interactions arising from ionic solvents in ionothermal routes,30 the role of Sr2+ should be taken into account for the controlled generation of TaON and Ta3N5 nanoparticles. | ||
| Fig. 4 XRD patterns of products obtained in the absence of Sr2+ with the varied RU/Ta ratio from 1.0 to 8.0. | ||
As the ionothermal processes were conducted at lower temperature (550 °C) or with a lower flow rate of carrier N2 (100 mL min−1) than that for a typical procedure (700 °C and 500 mL min−1), in which nitridation reactions were depressed,31 the intermediate of SrTa4O11 (ICDD no. 01-083-0608) was received (Fig. S5 in ESI†). This observation indicates the strong interactions between Sr and Ta in the form of strontium tantalate during ionothermal synthesis. According to the crystalline data of SrTa4O11 and Ta2O5 (Fig. S6 in ESI†), some weakened Ta–O bonds in SrTa4O11 are suggested by the extended length in a wider range (1.839–2.478 Å) than that in Ta2O5 (1.839–2.126 Å). This predicts the easier breaking of Ta–O in strontum tantalate under nitridation, and the promoted replacement of O atoms by N, as verified by the following thermal analysis.
The TGA/DSC analysis under N2 atmosphere was conducted on the similar gel precursors with and without Sr2+, which were denoted as SrTa/U and Ta/U, respectively (Fig. S7 in ESI†). The same RU/Ta of 2.0 was adopted. To elucidate the nitridation reactions, we focus on the evolution in TGA/DSC over 500 °C (Fig. 5a). Noticeably, the nitrogen source at such high temperature is CNx derived from urea,20,32 whose presence has been verified by IR spectra (Fig. S8 in ESI†). The SrTa/U precursor shows a weight loss and an endothermic peak around 626 °C, and another evolution near 765 °C. The samples taken from TGA/DSC at 650 and 800 °C respectively exhibit the typical absorption band-edges of TaON (490 nm) and Ta3N5 (600 nm) in UV-vis DRS (Fig. 5b). It is indicated that the reactions around 626 °C in TGA/DSC would be associated with the nitridation to TaON, at which point the further reaction to Ta3N5 requiring higher temperature of 765 °C is avoided. However, Ta/U without Sr2+ only presents a drastic weight loss and an obvious endothermic peak around 742 °C, consistent with the synthetic procedure requiring calcination at 775 °C (Fig. S9 in ESI†). And the sample taken from TGA/DSC at 800 °C displays an intensive absorption from 600 nm in UV-vis DRS (Fig. 5b), corresponding with the uncontrolled nitridation to TaON and Ta3N5 mixtures (Fig. 4). Similar situation is also observed in the case with a RU/Ta of 7.0 (Fig. S8 in ESI†). SrTa/U (RU/Ta = 7.0) obviously displays the accelerated nitridation at lower temperature than that of Ta/U due to the mediation of SrTa4O11. This effectively restricts the over-nitridation to TaN that is usually observed in the absence of Sr2+ (Fig. 4).
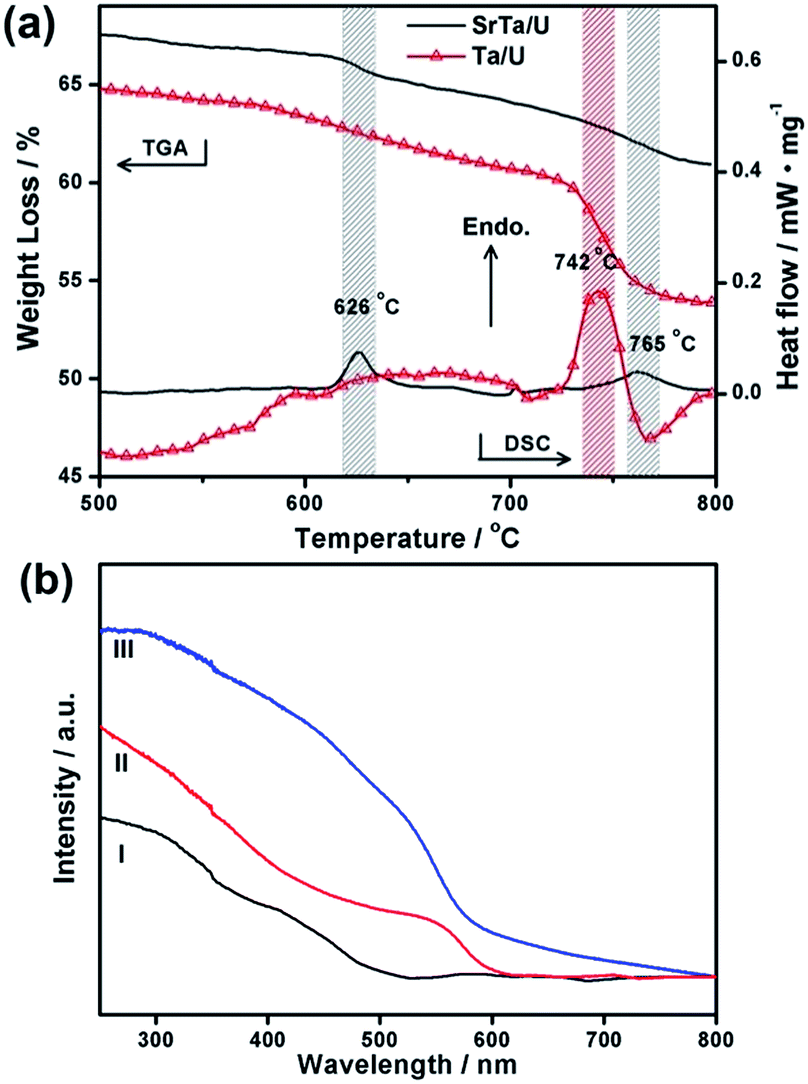 | ||
| Fig. 5 (a) TGA/DSC curves of SrTa/U and Ta/U with RU/Ta of 2.0, focusing on the evolution upon 500 °C. (b) UV-vis DRS of products taken from TGA/DSC analysis at (I) 650 and (II) 800 °C in SrTa/U (RU/Ta = 2.0), and at (III) 800 °C in Ta/U (RU/Ta = 2.0). | ||
Based on the above results, we propose a new mechanism associated with crucial strontium-tantalate intermediates for controlled nitridation (Scheme 1). In the intermediate of SrTa4O11, parts of Ta–O bonds are weakened. This benefits the replacement of O by N for generating TaON with a low RU/Ta, and more importantly avoids over-nitridation to Ta3N5 because the residual Ta–O bonds are not affected by Sr2+. In the case with a high RU/Ta, the promoted nitridation at T < 700 °C also prevents products from over-reacting to TaN. Thus, the controlled nitridation is successfully achieved. In this way, the Sr/Ta ratio controlling the intermediates of tantalates makes obvious influence the generation of TaON and Ta3N5 (Fig. S10 in ESI†).
 | ||
| Scheme 1 Proposed mechanism via a vital strontium-tantalate intermediate for controlled nitridation. | ||
This mechanism is universal for ionothermal processes using other alkaline-earth metal ions. In the presence of Ba2+, TaON and Ta3N5 phases are obtained as RU/Ta = 2.0 and 5.0, respectively (Fig. S11 in ESI†). Similar pure Ta3N5 is fabricated at RU/Ta = 5.0 with Ca2+ or Mg2+, however, the intermediates of Ca2Ta2O7 and MgTa2O6 still remain at the low RU/Ta of 1.0–3.0 (Fig. S12 and S13 in ESI†), whose decomposition usually requires complete nitridation with high temperature (>775 °C) and flow rate (>1.0 L min−1).28 Such difference is probably related to their diverse ionic radius (Mg2+ 89 pm; Ca2+ 112 pm; Sr2+ 126 pm; Ba2+ 142 pm).33 Owing to the toxicity of Ba2+ and the difficult decomposition of Ca2+ and Mg2+-based tantalates, the Sr2+-mediated route is suitable for the environment-friendly and facile synthesis of nitrides.
We believe that this explanation would make a progress in discovering the essential effects of ionic solvents. Ionothermal routes were ever conducted to fabricate ternary metal oxynitrides, e.g., MTaO2N (M = Ca, Sr or Ba), MNbO2N (M = Sr or Ba), however, their functions in controlled nitridation were unfortunately ignored.34 In our previous work,28 the indispensable effect of Ca2+ in the controlled synthesis of TaON and Ta3N5 was ascribed to the chelation between Ca2+ and urea, which slowed down the release of NH3 from urea decomposition to control nitride generation. However, it cannot elucidate how the urea decomposition at low temperature (<400 °C) affects nitridation at high temperature (∼775 °C). Herein, tantalate intermediates are found in the ionothermal routes with a wide range of alkaline-earth-metal salts (e.g., Mg2+, Ca2+, Sr2+ and Ba2+), which are indispensable to achieve the well-defined tantalum (oxy)nitrides. The controlled nitridation due to the weakened Ta–O bonds of tantalates successfully protects the desired generations of TaON and Ta3N5 from over-reactions, opening new mind in the understanding of ionothermal synthesis.
3.3 Metal-support interactions associated with controlled nitridation
Supported gold has recently emerged as versatile catalysts for a broad array of organic transformations, including a number of hydroprocessing reactions, in which the performance depends on its interactions with supports.13,14,35 Given the obviously varied electronic feature in tantalum (oxy)nitrides (cf. Fig. 3), metal-support interactions with gold nanocatalysts are expected to be tailored by controlled nitridation. In this regard, Au/Ta2O5, Au/TaON and Au/Ta3N5 with an Au loading of 0.8–0.9% were prepared via a same deposition–precipitation method. As shown in the TEM images (Fig. 6), Au nanoparticles with a uniform size around 3.5 nm are observed on the supports of Ta2O5, TaON and Ta3N5. | ||
| Fig. 6 TEM images of (a) 0.9% Au/Ta2O5, (c) 0.8% Au/TaON, (e) 0.9% Au/Ta3N5, and the corresponding size distribution of Au on (b) Ta2O5, (d) TaON, (f) Ta3N5. | ||
H2-TPR and XPS analysis were further conducted on the above catalysts. In H2-TPR results (Fig. 7a), the strong metal-support interactions in Au/TaON and Au/Ta3N5 are suggested by the lower reduction temperature of Au3+ species in comparison with Au/Ta2O5, all of which possess the consistent size-distribution of Au (Fig. 6). Although parallel reductions peaking at 121–122 °C are both detected, the onset temperature of 72 °C in Au/Ta3N5 is lower than that in Au/TaON (97 °C). Such interactions are further affirmed by XPS (Fig. 7b). The sample of Au/Ta2O5 displays the Au 4f7/2 and 4f5/2 peaks located at 87.6 and 83.9 eV, which are coincident with those for metallic gold.36 With the nitridation of Ta-based supports, these peaks are respectively shifted to the lower binding energies of 87.4 and 83.7 eV in Au/TaON, indicating negatively charged Au species (Auδ−). This observation should be attributed to the increased Fermi level in nitridated supports (Fig. S4b in ESI†), which promotes electron transfer from supports to Au.37–39 In Au/Ta3N5, the corresponding ones with an obvious red-shift to 87.1 and 83.4 eV suggest that electrons are further accumulated on Auδ− surface due to the highly nitridated Ta3N5 supports. Such metal-support interactions varied by the nitridation of Ta-based supports are expected to make obvious influence on the catalytic performance of gold.40,41
 | ||
| Fig. 7 (a) H2-TPR profiles and (b) Au 4f XPS spectra of 0.9% Au/Ta2O5, 0.8% Au/TaON, and 0.9% Au/Ta3N5. | ||
3.4 Nitrobenzene hydrogenation
The hydrogenation of nitrobenzene (NB), key routes towards important intermediates for dyestuff, pharmaceuticals, and food additives, is considered as an available probe reactions to identify the function of metal-support interactions due to its multiple steps and directions.42 In this work, NB hydrogenation was conducted over Au/Ta2O5, Au/TaON and Au/Ta3N5 using formic acid (FA) as a safe and economic hydrogen source.43 As shown in Table 1, phenylhydroxylamine (PHA) and aniline (AN) are the products, without nitrosobenzene or azobenzene being detected. A consecutive hydrogenation pathway is suggested, similar with that proposed on Au/TiO2 by Corma et al.42 All the supported Au catalysts show obviously higher activity than bare Ta-based nanoparticles (Entries 1–3 and 4–6 in Table 1), indicating Au as the predominant active species. The catalyst of 0.9% Au/Ta2O5 gives a low conversion of 52% with negligible AN selectivity (Entry 4 in Table 1). The improved activity with an almost complete conversion and a AN selectivity of 51% is achieved on 0.8% Au/TaON (Entry 5 in Table 1), which is superior to those on 0.9% Au/Ta3N5 and even on typical 0.8% Au/SiO2 (Entries 6 and 7 in Table 1). With other Au loading (0.5–2.0 wt%), the Au/TaON also presents a better activity than that on Au/Ta2O5 and Au/Ta3N5 (Table S2 in ESI†), suggesting a high activity associated with the TaON support. It can be further confirmed by NB reduction using H2, which shows analogous situation (Entries 8–10 in Table 1). Additionally, the AN selectivity on Au/TaON can be improved by increasing Au loading (Entries 5 and 11–13 in Table 1), agreeing with the nature of consecutive hydrogenation pathway.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Conv. (%) | Sel. (%) | |
| PHA | AN | |||
| a Typical conditions: NB (0.5 mmol), catalysts (40 mg), FA (6.0 mmol), EtOH (5.0 mL), H2O (5.0 mL), N2 (0.5 MPa), 80 °C, 2.5 hours.b H2 (0.5 MPa) was used as the reductant. | ||||
| 1 | Ta2O5a | 33 | >99 | 0 |
| 2 | TaONa | 30 | >99 | 0 |
| 3 | Ta3N5a | 35 | >99 | 0 |
| 4 | 0.9% Au/Ta2O5a | 52 | 92 | 8 |
| 5 | 0.8% Au/TaONa | >99 | 49 | 51 |
| 6 | 0.9% Au/Ta3N5a | 73 | 60 | 40 |
| 7 | 0.8% Au/SiO2a | 74 | 83 | 17 |
| 8 | 0.9% Au/Ta2O5b | 52 | >99 | 0 |
| 9 | 0.8% Au/TaONb | 65 | >99 | 0 |
| 10 | 0.9% Au/Ta3N5b | 58 | >99 | 0 |
| 11 | 0.5% Au/TaONa | 39 | 85 | 15 |
| 12 | 1.3% Au/TaONa | >99 | 29 | 71 |
| 13 | 1.7% Au/TaONa | >99 | 27 | 73 |

Considering the similar catalytic FA decomposition to CO2 and H2 over Au/Ta2O5, Au/TaON and Au/Ta3N5 (Table S3 in ESI†), the different performance for NB reduction by FA should be contributed by the reduction steps of NB. Further test in the direct hydrogenation of PHA exhibits the negligible difference (Table S4 in ESI†), suggesting the varied activity in the first step from NB to PHA. The intrinsic properties of Au in the above catalysts are believed as a key factor for NB hydrogenation, regarding the similarities in surface area, Au particle size and active-site amount already confirmed by N2 sorption analysis, TEM investigation and CO uptake experiment, respectively (Table S1 in ESI† and Fig. 6).
Typically, the adsorption of NB substrate on Au is the key in NB reduction.44,45 According to the Balandin-type volcano plots, the moderate adsorption on metals is demanded for efficient catalytic conversion because a weak adsorption cannot effectively activate substrates and a strong adsorption precludes product desorption.46,47 Negatively charged Auδ− over TaON resulting from electronic metal-support interactions38,39 (cf. XPS in Fig. 7b) would promote the adsorption of polar –Nδ+–Oδ− bonds in NB,42,46,47 as compared with the neutral Au on Ta2O5, which presents optimized activity and selectivity (Scheme 2). As for Au/Ta3N5, the further negatively charged Auδ− on Ta3N5 would make NB and even as-formed PHA adsorption so strong that the following steps are hindered.46 And thus, the reduced activity and AN selectivity are observed. Obviously, an appropriate nitridation level of supports is critical for optimizing metal-support interactions with gold nanocatalysts.
 | ||
| Scheme 2 Schematic illustration for NB hydrogenation over Au/Ta2O5, Au/TaON and Au/Ta3N5. | ||
Another test under light irradiation, showing further affected charge-distribution on Au by photo-excited electrons, can serve as an important evidence for the above conclusion in nonphotocatalytic condition. In view of the different band-gap energy of Ta2O5, TaON and Ta3N5, UV (λ > 200 nm) and visible (λ = 400–800 nm) light was employed for Au/Ta2O5 and the both of Au/TaON and Au/Ta3N5, respectively. The increased activity is observed on Au/Ta2O5, while the decreased one is presented by Au/TaON and Au/Ta3N5, in contrast with that without irradiation (Fig. 8). Thanks to the excited electron from Ta2O5 to Au, the as-formed Auδ− accelerates NB reduction by enhanced adsorption. However, too strong NB and PHA adsorption on excessively charged Auδ−, strengthened by photo-excitation, would block the following reactions and thus reduce the activity on Au/TaON and Au/Ta3N5. Additionally, the test under visible light (λ = 400–800 nm) was also conducted on Au/Ta2O5. The performance similar to that without irradiation, but lower than that with UV, should be ascribed to the failure to excite wide-band-gap Ta2O5 (3.9 eV) by visible light. This test eliminates the influence from the visible-light plasmonic effects of Au.
 | ||
| Fig. 8 Comparison of NB hydrogenation over Ta-NPs supported Au with and without light irradiation. | ||
4. Conclusions
A Sr2+-mediated ionothermal synthesis with an improved mechanism has been successfully introduced to fabricate tantalum (oxy)nitride nanoparticles with well-defined composition and electronic properties. This work demonstrates a strategy to vary the electronic features of tantalum (oxy)nitrides towards optimized metal-support interactions with Au, which promotes the hydrogenation of NB by controlling the electronic feature of Au and the consequent substrate adsorption/activation. Our effort highlights the advantages of controlled nitridation for achieving optimized metal-support interactions, pointing out a new protocol for catalyst design.Acknowledgements
This work is financially supported by the National Basic Research Program of China (2013CB934101), National Natural Science Foundation of China (21203075, 21373102, and 21433002) and Fundamental Research Funds for the Central Universities (21615402). Q. S. Gao also thanks the support from Guangdong Natural Science Funds (2015A030306014 and 2014TQ01N036) and Guangdong Higher Education Institute (YQ2013022).Notes and references
- A. Salamat, A. L. Hector, P. Kroll and P. F. McMillan, Coord. Chem. Rev., 2013, 257, 2063–2072 CrossRef CAS PubMed.
- J. S. J. Hargreaves, Coord. Chem. Rev., 2013, 257, 2015–2031 CrossRef CAS PubMed.
- X. J. Lang, X. D. Chen and J. C. Zhao, Chem. Soc. Rev., 2014, 43, 473–486 RSC.
- N. E. Breese, Structure and Bonding, Crystal Chemistry of Inorganic Nitride, Springer, Berlin, 1992 Search PubMed.
- E. Furimsky, Appl. Catal., A, 2003, 240, 1–28 CrossRef CAS.
- V. Heine, Phys. Rev., 1967, 153, 673–682 CrossRef CAS.
- A. M. Alexander and J. S. J. Hargreaves, Chem. Soc. Rev., 2010, 39, 4388–4401 RSC.
- Q. S. Gao, N. Liu, S. N. Wang and Y. Tang, Nanoscale, 2014, 6, 14106–14120 RSC.
- M. M. O. Thotiyl, T. R. Kumar and S. Sampath, J. Phys. Chem. C, 2010, 114, 17934–17941 CAS.
- V. Molinari, C. Giordano, M. Antonietti and D. Esposito, J. Am. Chem. Soc., 2014, 136, 1758–1761 CrossRef CAS PubMed.
- N. Perret, F. Cardenas-Lizana, D. Lamey, V. Laporte, L. Kiwi-Minsker and M. A. Keane, Top. Catal., 2012, 55, 955–968 CrossRef CAS.
- F. Cardenas-Lizana, D. Lamey, N. Perret, S. Gomez-Quero, L. Kiwi-Minsker and M. A. Keane, Catal. Commun., 2012, 21, 46–51 CrossRef CAS PubMed.
- X. Liu, L. He, Y. M. Liu and Y. Cao, Acc. Chem. Res., 2014, 47, 793–804 CrossRef CAS PubMed.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- X. H. Li and M. Antonietti, Chem. Soc. Rev., 2013, 42, 6593–6604 RSC.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851–7861 CAS.
- R. Abe, M. Higashi and K. Domen, J. Am. Chem. Soc., 2010, 132, 11828–11829 CrossRef CAS PubMed.
- Y. G. Su, J. Y. Lang, L. P. Li, K. Guan, C. F. Du, L. M. Peng, D. Han and X. J. Wang, J. Am. Chem. Soc., 2013, 135, 11433–11436 CrossRef CAS PubMed.
- Q. S. Gao, S. N. Wang, Y. C. Ma, Y. Tang, C. Giordano and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 961–965 CrossRef CAS PubMed.
- Q. S. Gao, C. Giordano and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 11740–11744 CrossRef CAS PubMed.
- M. E. Strayer, J. M. Binz, M. Tanase, S. M. K. Shahri, R. Sharma, R. M. Rioux and T. E. Mallouk, J. Am. Chem. Soc., 2014, 136, 5687–5696 CrossRef CAS PubMed.
- A. Kumar and V. Ramani, ACS Catal., 2014, 4, 1516–1525 CrossRef CAS.
- K. An, S. Alayoglu, N. Musselwhite, K. Na and G. A. Somorjai, J. Am. Chem. Soc., 2014, 136, 6830–6833 CrossRef CAS PubMed.
- E. Orhan, F. Tessier and R. Marchand, Solid State Sci., 2002, 4, 1071–1076 CrossRef CAS.
- M. Kerlau, O. Merdrignac-Conanec, M. Guilloux-Viry and A. Perrin, Solid State Sci., 2004, 6, 101–107 CrossRef CAS PubMed.
- C. Giordano and M. Antonietti, Nano Today, 2011, 6, 366–380 CrossRef CAS PubMed.
- Q. S. Gao, C. Giordano and M. Antonietti, Small, 2011, 7, 3334–3340 CrossRef CAS PubMed.
- W. J. Chun, A. Ishikawa, H. Fujisawa, T. Takata, J. N. Kondo, M. Hara, M. Kawai, Y. Matsumoto and K. Domen, J. Phys. Chem. B, 2003, 107, 1798–1803 CrossRef CAS.
- X. F. Liu, N. Fechler and M. Antonietti, Chem. Soc. Rev., 2013, 42, 8237–8265 RSC.
- Z. Schnepp, M. Thomas, S. Glatzel, K. Schlichte, R. Palkovits and C. Giordano, J. Mater. Chem., 2011, 21, 17760–17764 RSC.
- D. Mitoraj and H. Kisch, Chem.–Eur. J., 2010, 16, 261–269 CrossRef CAS PubMed.
- J. G. Speight, Lange's Handbook of Chemistry, McGraw-Hill Professional, New York, 16th edn, 2005 Search PubMed.
- A. Gomathi, S. Reshma and C. N. R. Rao, J. Solid State Chem., 2009, 182, 72–76 CrossRef CAS PubMed.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- P. Kast, G. Kucerova and R. J. Behm, Catal. Today, 2015, 244, 146–160 CrossRef CAS PubMed.
- Y.-N. Sun, L. Giordano, J. Goniakowski, M. Lewandowski, Z.-H. Qin, C. Noguera, S. Shaikhutdinov, G. Pacchioni and H.-J. Freund, Angew. Chem., Int. Ed., 2010, 49, 4418–4421 CrossRef CAS PubMed.
- X. Y. Liu, A. Q. Wang, T. Zhang and C. Y. Mou, Nano Today, 2013, 8, 403–416 CrossRef CAS PubMed.
- A. M. Kolpak, I. Grinberg and A. M. Rappe, Phys. Rev. Lett., 2007, 98, 166101 CrossRef.
- T. Mitsudome and K. Kaneda, Green Chem., 2013, 15, 2636–2654 RSC.
- S. Schauermann, N. Nilius, S. Shaikhutdinov and H. J. Freund, Acc. Chem. Res., 2013, 46, 1673–1681 CrossRef CAS PubMed.
- A. Corma, P. Concepcion and P. Serna, Angew. Chem., Int. Ed., 2007, 46, 7266–7269 CrossRef CAS PubMed.
- I. Sorribes, G. Wienhofer, C. Vicent, K. Junge, R. Llusar and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 7794–7798 CrossRef CAS PubMed.
- L. Y. Zou, Y. Y. Cui and W. L. Dai, Chin. J. Chem., 2014, 32, 257–262 CrossRef CAS PubMed.
- M. Boronat, P. Concepcion, A. Corma, S. Gonzalez, F. Illas and P. Serna, J. Am. Chem. Soc., 2007, 129, 16230–16237 CrossRef CAS PubMed.
- M. Turakova, T. Salmi, K. Eraenen, J. Waerna, D. Y. Murzin and M. Kralik, Appl. Catal., A, 2015, 499, 66–76 CrossRef CAS PubMed.
- L. Zhang, J. Jiang, W. Shi, S. Xia, Z. Ni and X. Xiao, RSC Adv., 2015, 5, 34319–34326 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization of SEM, TEM, XRD, TGA/DSC, IR, N2 isothermal sorption and CO uptake. See DOI: 10.1039/c5ra19644b |
| This journal is © The Royal Society of Chemistry 2015 |