
Fe/N co-doped carbon microspheres as a high performance electrocatalyst for the oxygen reduction reaction†
Abstract
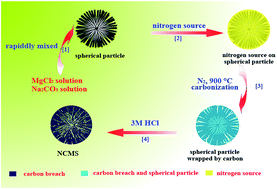
Recently, nitrogen-doped carbon materials have attracted immense interest because of their great potential in various applications. In this work, FeN-doped carbon microspheres are large-scale synthesized using basic magnesium carbonate as the template and glycine as the carbon and nitrogen precursor by a simple liquid impregnation method under a relatively low pyrolysis temperature. Iron is introduced into the carbon microspheres to enhance the graphitic degree and improve the electrocatalytic performance. This carbon material with high specific area, high nitrogen content and part-graphitization shows high activity and four-electron selectivity for the oxygen reduction reaction in an alkaline medium. Compared to a commercial Pt/C catalyst, this material presents exceeding stability and durability, which can be a candidate for potential applications in the fuel cell and electrochemical industries of oxygen reduction.
 Please wait while we load your content...
Please wait while we load your content...

