
Molecular orientation within thin films of isomorphic molecular semiconductors†
Abstract
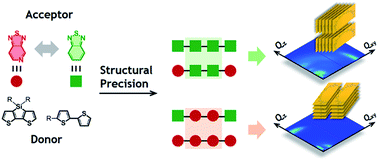
Four isomorphic organic semiconductors are compared to map out how the precision of chemical structures determines solid state molecular organization. Intramolecular electronic structure and intermolecular packing preference in the solid state were shown to be exclusively dependent on the relative location of the electron accepting fragments within a given molecule.
 Please wait while we load your content...
Please wait while we load your content...

