
Role of polar solvents for the synthesis of pillar[6]arenes†
S. Santraa,
I. S. Kovaleva,
D. S. Kopchukab,
G. V. Zyryanovab,
A. Majeec,
V. N. Charushinab and
O. N. Chupakhinab
aUral Federal University, Chemical Engineering Institute, Yekaterinburg, 620002, Russian Federation. E-mail: gvzyryanov@gmail.com
bI. Ya. Postovskiy Institute of Organic Synthesis, Ural Division of the Russian Academy of Sciences, 22 S. Kovalevskoy Str., Yekaterinburg, 620219, Russian Federation. E-mail: chupakhin@ios.uran.ru
cDepartment of Chemistry, Visva-Bharati (A Central University), Santiniketan-731235, India. E-mail: adinath.majee@visva-bharati.ac.in
First published on 1st December 2015
Abstract
An efficient procedure for the synthesis of pillar[6]arenes has been developed. The procedure involves the condensation of 1,4-dialkoxybenzenes and paraformaldehyde in the presence of a catalytic amount of H2SO4 or BF3·OEt2 in polar solvent media (acetonitrile, ethyl alcohol, acetone etc.). In all cases the interaction afforded pillar[6]arenes in high yields.
Pillar[n]arenes (n = 5 (ref. 1) or 6 (ref. 2)) are called “a fascinating cyclophanes with a bright future”.3 Indeed, these macromolecules are quite prospective molecular cavity containers due to their rigid penta- or hexagon shape and high-electron rich cavity of 5.5–7.5 Å diameter.1,4 Pillararenes have wide applications in supramolecular chemistry as supramolecular hosts and tectons,4 in analytical chemistry as sensor molecules,5 for instance, the colorimetric detection of metal cations,6 fluorescent7 or electrochemical8 detection of nitrogen-based cations, as well as anions sensing9 and some neutral molecules binding/encapsulation.10 In material science pillararenes have been used as components in liquid crystals,11 photoresists,12 ion-selective membranes,13 as artificial gas14 and water15 channels. Some important biological applications of pillar[5]arenes have also been reported.16
Pillar[6]arenes (and higher pillar[n]arenes) are commonly isolated as by-products in the synthesis of pillar[5]arenes, which involves the Lewis or Brønsted acid-catalyzed cyclocondensation between paraformaldehyde and 1,4-dialkyloxybenzenes in chlorinated solvents (Scheme 1, way i).1 In fact, the formation of pillar[5]arenes in the most cases occurs under thermodynamic control, therefore yields of higher pillar[n]arenes, the kinetic products, are normally low.17a–b Only in one report Cao, Meier and co-authors carried out the synthesis of pillar[6]arenes under the kinetically controlled conditions by using FeCl3 in anhydrous chloroform to yield the mixture of pillar[5]- and pillar[6]arenes with the preferable formation of larger macrocycle in the ratios, depending on the nature of alkyl substituents and the reaction time.17c Other approaches, including the Brønsted acid-catalyzed self-condensation of bis(alkoxymethyl)-1,4-dialkoxybenzene2,17d (way ii) or Lewis acid-promoted self-condensation 2,5-alkoxybenzyl alcohols (way iii),2,17e–f reported by the Meier's and Huang's research groups afforded only the minor amounts of less stable pillar[6]arenes. Recently, Ogoshi and co-authors observed the formation of some pillar[6]arenes in high yields using 1-chlorocyclohexane as reaction media and as a template.19a Very recently, the Zhang group developed a method for the synthesis of pillar[6]arene with 53% yield by condensation of 1,4-dialkoxybenzene and paraformaldehyde with the choline chloride (ChCl)/ferric chloride (FeCl3) deep eutectic solvent in CH2Cl2 at room temperature.20
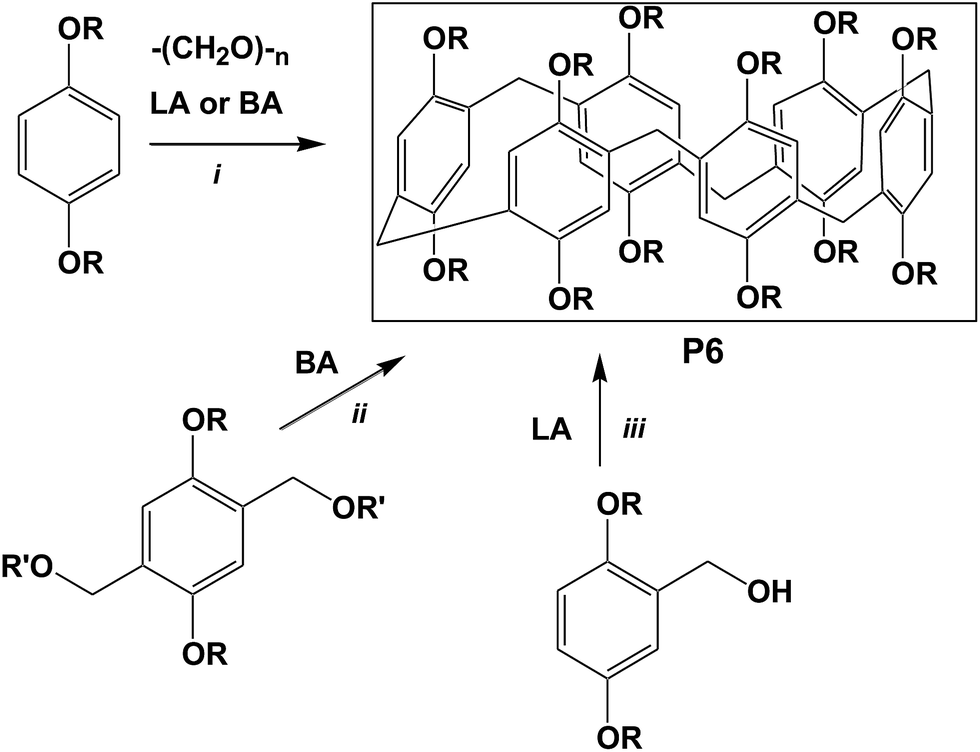 | ||
| Scheme 1 Common approaches to the synthesis of pillar[6]arenes. | ||
All these methodologies reported so far for synthesis of pillar[6]arenes furnished very poor yields and use of chlorinated solvent is another drawback. So, high-yield synthesis in non-chlorinated and/or polar solvents is demanding for current working practice.
In continuation of our research in organic methodologies we have reported very recently a very clean synthesis of pillar[6]arenes18 in absence of solvent. To investigate the role of solvent we have examined the synthesis of pillar[6]arenes (including one novel compound) in acetonitrile and other common polar solvents in the presence of catalytic amount of sulphuric acid or BF3·OEt2.
Results and discussion
According to Ogoshi and co-authors19a the role of the non-polar chlorinated solvent in a synthesis of pillar[n]arenes is in the template effect, promoting the formation of the desired pillar[n]arene (mostly n = 5), depending on the geometric size of the solvent. Thus, in the classical Ogoshi's procedure1 and its following modifications21 linear dichloroalkanes occupy the cavity of pillar[5]arene to promote the preferable formation of more stable pillar[5]arenes over pillar[6]arenes during the thermodynamically controlled cyclisation reaction. In the absence of linear dichloroalkanes, for instance in chloroform,17b,c under the kinetic control the reaction led to the mixture of pillar[n]arenes with the preferable formation of pillar[6]arenes and larger macrocycles which are kinetic products, and upon standing their following interconversion to pillar[5]arenes and/or linear oligomers occurs. If the solvent for the reaction media has a proper size to template the pillar[6]arene; this macrocycle was the only product due to its “internal locking” by the template solvent. For instance, Ogoshi and co-authors isolated pillar[6]arenes as major products using 1-chlorocyclohexane as a solvent.19a The preferable formation of pillar[5]arenes in solvents other than 1-chlorocyclohexane is based on mechanistic aspects of this reaction as a Friedel–Crafts alkylation.Thus, according to the literature,22 on the first step the acid-catalyzed hydroxyl methylation reaction of 1,4-dialkoxy benzene with paraformaldehyde (or in some cases formaldehyde22a or 1,3,5-trioxane23) afforded the benzyl alcohol A (Scheme 2). Elimination of water molecule from the benzyl alcohol A converts it to a benzylic cation B. The benzylic cation under acidic conditions underwent further reaction with another molecule of benzyl alcohol to give a dimeric benzylic cation C, and finally other oligomeric cations D. At the final step, the oligomeric cation D stabilized through the cyclization to the most stable pillar[n]arene, i.e. pillar[5]arene P5.
 | ||
| Scheme 2 The proposed mechanism for the formation of pillar[n]arenes. | ||
On the other hand Neumann and co-authors24 described a high yield method for the preparation of aromatic aldehydes via the oxidative hydroxymethylation reaction of various arenes by paraformaldehyde in acetonitrile catalyzed by H2SO4 in the presence of oxidant (DDQ). In this reaction the in situ formed benzyl alcohol quantitatively converts into the corresponding aldehyde. Surprisingly, the authors only mentioned the possibility for the formation of higher linear diarylmethanes as by-products under the hydroxymethylation conditions if no oxidant would present.
Keeping in mind that pillar[n]arenes have been efficiently obtained by the Friedel–Crafts alkylation reaction in chlorinated solvents, we became interested in developing of other synthetic procedures to pillar[n]arenes,‡ for instance based on the modified Neumann's procedure, i.e. in oxidant-free conditions.
The outcome of the reaction was quite unexpected. Effectively, no linear oligomeric compounds or pillar[5]arene could be isolated and the pillar[6]arene 2a was the major cyclized product. As shown in Scheme 3, treatment of 1,4-diethoxybenzene (1a) with H2SO4 in acetonitrile at room temperature gave pillar[6]arene 2a. Meanwhile, other 1,4-dialkoxybenzenes (except R = Bn) under this conditions afforded pillar[6]arenes 2b–d in good yields (Table 1). Moreover, this present methodology is also applicable for synthesizing ester substituted pillar[6]arene (2f) with moderate yield.
 | ||
| Scheme 3 Synthesis of pillar[6]arenes 2a–f in polar solvents media. | ||
The yields of pillar[6]arenes 2 are improved significantly over those previously reported for the synthesis of pillar[6]arenes (36–53%, except the recent Ogoshi's report). The optimization of the reaction conditions is shown in Table 2. The optimum ratio of 1,4-diethoxybenzene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :paraformaldehyde:H2SO4 is 1:2:0.3 (30% molar) (entry 3). The increasing of the amount of paraformaldehyde did not improve the yield of 2a significantly. The variation of the amount of H2SO4 (entries 6–8) resulted in the decreasing of the yield of 2a. Probably, use of large amount of H2SO4 results the formation of insoluble long chain polymer. The reaction is completed normally within 5 minutes, and the prolonging of the reaction time decreased the yield of the target product (entries 9 and 10).
:paraformaldehyde:H2SO4 is 1:2:0.3 (30% molar) (entry 3). The increasing of the amount of paraformaldehyde did not improve the yield of 2a significantly. The variation of the amount of H2SO4 (entries 6–8) resulted in the decreasing of the yield of 2a. Probably, use of large amount of H2SO4 results the formation of insoluble long chain polymer. The reaction is completed normally within 5 minutes, and the prolonging of the reaction time decreased the yield of the target product (entries 9 and 10).
| Entry | Amount of PF used (equiv.) | Amount of H2SO4 | Timeb [min] | Yields of 2ac [%] |
|---|---|---|---|---|
| a All reactions were carried out on 2 mmol scale in 2 mL of acetonitrile.b Time was counted after addition of the catalyst.c Yields after washing with ethyl alcohol. PF = paraformaldehyde. | ||||
| 1 | 1 | 30 mol% | 5 | 53 |
| 2 | 1.5 | 30 mol% | 5 | 61 |
| 3 | 2 | 30 mol% | 5 | 71 |
| 4 | 2.5 | 30 mol% | 5 | 69 |
| 5 | 3 | 30 mol% | 5 | 68 |
| 6 | 2 | 50 mol% | 5 | 51 |
| 7 | 2 | 10 mol% | 5 | 31 |
| 8 | 2 | 20 mol% | 5 | 55 |
| 9 | 2 | 30 mol% | 10 | 68 |
| 10 | 2 | 30 mol% | 15 | 62 |
Surprisingly, similar results were observed in acetonitrile in the presence of the Lewis acid, i.e. BF3·OEt2, which is the most common for the pillar[5]arene synthesis: the pillar[6]arene 2a was isolated in up to 67% yield (see Table 1, ESI†).
Similarly to the previously reported procedures the reaction proceeds via the in situ formation of benzyl alcohols. To prove that we synthesized benzyl alcohols 4a, 4b and their following condensation in acetonitrile in the presence of H2SO4 afforded pillar[6]arenes 2a, 2b in high yields (see Scheme 1, ESI†).
To investigate further the influence of the nature and the polarity of solvents on the selective formation of pillar[6]arene, we examined several other solvents and results are presented in Table 3. According to the experimental data in the media of most polar solvents with a high dipole moment (entries 1–7) the pillar[6]arene 2a was obtained in 55–72% yields depending on catalyst. The decreasing of the dipole moment of solvents resulted in the lower yields of 2a (see Fig. 1, ESI†). Some common solvents with low polarity such as ethyl cyanoacetate, nitromethane, chloroacetonitrile, acetone and ethyl alcohol also served as good solvents to produce pillar[6]arene 2a (entries 4–8).
| Entry | Solvent | Dipole momentc [D] | Yield [%] |
|---|---|---|---|
| a All reactions were carried out on 2 mmol scale in different solvents.b Based on 1H NMR data after workup.c Dipole moment values are published elsewhere.d BF3·OEt2 used as a catalyst.e The 1,4-diethoxybenzene did not dissolve properly.f Dipole moment values were calculated using DFT (B3LYP 6-311G) calculations.g The addition of H2SO4 caused the polymerisation of the reaction mixture.h From ref. 17c.i From ref. 17b. | |||
| 1 | Benzonitrile | 4.51 | 72(63)d |
| 2 | Nitrobenzene | 4.28 | 71 (70)d |
| 3 | Acetonitrile | 3.92 | 71(67)d |
| 4 | Ethyl cyanoacetate | 3.80 | 67(60)d |
| 5 | Nitromethane | 3.56 | 61 |
| 6 | Chloroacetonitrile | 3.00 | 58 |
| 7 | Acetone | 2.88 | 55 |
| 8 | Ethyl alcohol | 1.69 | 43(52)d |
| 9 | Benzyl cyanide | N/A | 0e |
| 10 | Dichloroacetonitrile | 2.33f | N/A |
| 11 | Trichloroacetonitrile | 1.38f | 0g |
| 12 | 1,2-Dichloroethane | 1.80 | Traces |
| 13 | Chloroform | 1.15 | 34h(15)i |
| 14 | Dichloromethane | 1.14 | Traces |
 | ||
| Fig. 1 The possible role of polar solvent in the synthesis of pillar[6]arenes. | ||
Finally, when we reproduced the classical Ogoshi's procedure1,21b in a media of neat 1,2-dichloroethane, neat acetonitrile or in a 1:1 mixture of both solvents the resulting pillar[6]arene 2a was isolated in 0%, 67%, and 63% yields respectively. Similar results observed in the presence of H2SO4 (see Table 2, ESI†).
The possible role of polar solvents in this reaction is both the stabilization and the “external blocking” of in situ formed oligomeric cation (Fig. 1, top) and the following “external blocking”/templating of the cyclic cation (Fig. 1, middle) and the target hexamer (Fig. 1, bottom).22,25
The larger the dipole moment of solvent the stronger the oligomeric cation/dipole interactions, causing the more efficient charge distribution in a solute cation and the better stabilization of the cation due to the solvent effects,26 the higher its conversion into pillar[6]arene 2a. In some cases at the certain point the precipitation and further isolation of pillar[6]arene 2a from the reaction media may occur. All of these factors prevent further the interconversion of pillar[6]arenes into pillar[5]arenes, larger pillararenes or linear oligomers/polymers, which was possible during the preparation of pillararenes under the Friedel–Crafts conditions in chlorinated non-polar solvents due to the “dynamic character of covalent bond formation”.22a
This polar-solvent protocol is also applicable on a gram-scale synthesis. Upon ten times increasing of the amount of reagents in the presence H2SO4 or BF3·OEt2 in acetonitrile solution the yield of 2a was 69% and 64% respectively (see Scheme 2, ESI†).
Conclusions
In conclusion, we have demonstrated that H2SO4 in acetonitrile and other polar solvents media can serve as a robust and efficient catalytic system for the preparation of pillar[6]arenes. Due to the simplicity and practicability of both starting materials and the catalytic system, our findings would be expected to bring new insights in the research field of pillar[n]arene-based supramolecular chemistry and material science. The reaction was found to afford mainly pillar[6]arenes in a media of other common polar solvents and/or common Lewis acids.Acknowledgements
This work was supported by the Russian Science Foundation (Ref. # 15-13-10033) and, for A. M, by BRNS–DAE (Ref. No. 37(2)/14/35/2014-BRNS/563, June 10, 2014).Notes and references
- T. Ogoshi, S. Kanai, S. Fujinami, T.-A. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed.
- D. Cao, Y. Kou, J. Liang, Z. Chen, L. Wang and H. Meier, Angew. Chem., 2009, 121, 9901–9903 (Angew. Chem., Int. Ed., 2009, 48, 9721–9723) CrossRef.
- P. J. Cragg and K. Sharma, Chem. Soc. Rev., 2012, 41, 597–607 RSC.
- (a) T. Ogoshi and T. Yamagishi, Eur. J. Org. Chem., 2013, 2961–2975 CrossRef CAS; (b) W. Si, L. Chen, X.-B. Hu, G. Tang, Z. Chen, J.-L. Hou and Z.-T. Li, Angew. Chem., Int. Ed., 2011, 50, 12564–12568 CrossRef CAS; (c) H. Zhang, K. T. Nguyen, X. Ma, H. Yan, J. Guo, L. Zhu and Y. Zho, Org. Biomol. Chem., 2013, 11, 2070–2074 RSC; (d) X. Yan, P. Wei, Z. Li, B. Zheng, S. Dong, F. Huang and Q. Zhou, Chem. Commun., 2013, 49, 2512–2514 RSC; (e) N. L. Strutt, H. Zhang, M. A. Giesener, J. Lei and J. A. Stoddart, Chem. Commun., 2012, 48, 1647–1649 RSC; (f) S. Dong, J. Yuan and F. Huang, Chem. Sci., 2014, 5, 247–252 RSC. For pillararene-based MOFs see: (g) N. L. Strutt, D. Fairen-Jimenez, J. Iehl, M. B. Lalonde, R. Q. Snurr, O. K. Farha, J. T. Hupp and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 19136–19145 CrossRef PubMed; for reviews see: (h) T. Ogoshi and T. Yamagishi, Chem. Commun., 2014, 50, 4776–4787 RSC; (i) T. Ogoshi and T. Yamagishi, Bull. Chem. Soc. Jpn., 2013, 86, 312–332 CrossRef CAS; (j) D. Cao and H. Meier, Asian J. Org. Chem., 2014, 3, 244–262 CrossRef CAS.
- R. R. Kothur, B. A. Patel and P. J. Cragg, ScienceJet, 2015, 4, 1–8 CrossRef , and references therein.
- Y. M. Jia, Y. Y. Fang, Y. Li, L. T. He, W. H. Fan, W. Feng, Y. Y. Yang, J. L. Liao, N. Liu and L. H. Yuan, Talanta, 2014, 125, 322–328 CrossRef CAS.
- (a) N. L. Strutt, R. S. Forgan, J. M. Spruell, Y. Y. Botros and J. A. Stoddart, J. Am. Chem. Soc., 2011, 133, 5668–5671 CrossRef CAS PubMed; (b) P. Wang, Y. Yao and M. Xue, Chem. Commun., 2014, 50, 5064–5067 RSC.
- J. Zhou, M. Chen, J. Xie and G. Diao, ACS Appl. Mater. Interfaces, 2013, 5, 11218–11224 CAS.
- (a) S. Sun, X. Y. Hu, D. Z. Chen, J. B. Shi, Y. P. Dong, C. Lin, Y. Pan and L. Wang, Polym. Chem., 2013, 4, 2224–2229 RSC; (b) G. C. Yu, Z. B. Zhang, C. Y. Han, M. Xue, Q. Z. Zhou and F. Huang, Chem. Commun., 2012, 48, 2958–2960 RSC.
- (a) G. Yu, C. Han, Z. Zhang, J. Chen, X. Yan, B. Zheng, S. Liu and F. Huang, J. Am. Chem. Soc., 2012, 134, 8711–8717 CrossRef CAS PubMed; (b) Y. Kou, H. Tao, D. Cao, Z. Fu, D. Schollmeyer and H. Meier, Eur. J. Org. Chem., 2010, 6464–6470 CrossRef CAS; (c) X. Shu, S. Chen, J. Li, Z. Chen, L. Weng, X. Jia and C. Li, Chem. Commun., 2012, 48, 2967–2969 RSC. For hexanes: (d) Z. Zhang, B. Xia, C. Han, Y. Yu and F. Huang, Org. Lett., 2010, 12, 4360–4363 CrossRef. For solvents: (e) L.-L. Tan, Y. Zhang, B. Li, K. Wang, S. X.-A. Zhang, Y. Tao and Y.-W. Yang, New J. Chem., 2014, 38, 845–851 RSC.
- (a) I. Nierengarten, S. Guerra, M. Holler, J.-F. Nierengarten and R. Deschenaux, Chem. Commun., 2012, 48, 8072–8074 RSC; (b) I. Nierengarten, S. Guerra, M. Holler, L. Karmazin-Brelot, J. Barberá, R. Deschenaux and J.-F. Nierengarten, Eur. J. Org. Chem., 2013, 3675–3684 CrossRef CAS.
- H. Yamamoto, H. Kudo and T. Kozawa, Proc. SPIE 9051, Advances in Patterning Materials and Processes XXXI, 2014, p. 90511Z, DOI:10.1117/12.2046595.
- (a) R. R. Kothur, J. Hall, B. A. Patel, C. L. Leong, M. G. Boutelle and P. J. Cragg, Chem. Commun., 2014, 50, 852–854 RSC; (b) Q. Zhao, J. W. C. Dunlop, X. Qiu, F. Huang, Z. Zhang, J. Heyda, J. Dzubiella, M. Antonietti and J. Yuan, Nat. Commun., 2014, 5, 4293, DOI:10.1038/ncomms5293.
- (a) L.-L. Tan, H. Li, Y. Tao, S. X.-A. Zhang, B. Wang and Y.-W. Yang, Adv. Mater., 2014, 26, 7027–7031 CrossRef CAS PubMed; (b) T. Ogoshi, R. Sueto, K. Yoshikoshi and T. Yamagishi, Chem. Commun., 2014, 50, 15209–15211 RSC. For Xe-gas storage see: (c) T. Adiri, D. Marciano and Y. Cohen, Chem. Commun., 2013, 49, 7082–7084 RSC.
- (a) X. B. Hu, Z. Chen, G. Tang, J. L. Hou and Z. T. Li, J. Am. Chem. Soc., 2012, 134, 8384–8387 CrossRef CAS PubMed; (b) M. Barboiu, Angew. Chem., Int. Ed., 2012, 51, 11674–11676 CrossRef CAS PubMed.
- (a) C. Li, J. Ma, L. Zhao, Y. Zhang, Y. Yu, X. Shu, J. Li and X. Jia, Chem. Commun., 2013, 49, 1924–1926 RSC; (b) D.-D. Zheng, D.-Y. Fu, Y. Wu, Y.-L. Sun, L.-L. Tan, T. Zhou, S.-Q. Ma, X. Zha and Y.-W. Yang, Chem. Commun., 2014, 50, 3201–3203 RSC.
- (a) K. Wang, L.-L. Tan, D.-X. Chen, N. Song, G. Xi, S. X.-A. Zhang, C. Li and Y.-W. Yang, Org. Biomol. Chem., 2012, 10, 9405–9409 RSC; (b) X.-B. Hu, Z. Chen, L. Chen, L. Zhang, J.-L. Hou and Z.-T. Li, Chem. Commun., 2012, 48, 10999–11001 RSC; (c) H. Tao, D. Cao, L. Liu, Y. Kou, L. Wang and H. Meier, Sci. China: Chem., 2012, 55, 223–228 CrossRef CAS; (d) Y. Kou, D. Cao, H. Tao, L. Wang, J. Liang, Z. Chen and H. Meier, J. Inclusion Phenom. Macrocyclic Chem., 2013, 77, 279–289 CrossRef CAS; (e) Y. Ma, Z. Zhang, X. Ji, C. Han, J. He, Z. Abliz, W. Chen and F. Huang, Eur. J. Org. Chem., 2011, 5331–5335 CrossRef CAS; (f) C. Han, F. Ma, Z. Znang, B. Xia, Y. Yu and F. Huang, Org. Lett., 2010, 12, 4360–4363 CrossRef CAS PubMed.
- S. Santra, D. S. Kopchuk, I. S. Kovalev, G. V. Zyryanov, A. Majee, V. N. Charushin and O. N. Chupakhin, Green Chem., 2015 10.1039/c5gc01505g , Advance Article.
- (a) T. Ogoshi, N. Ueshima, T. Akutsu, D. Yamafuji, T. Furuta, F. Sakakibaraa and T. Yamagishi, Chem. Commun., 2014, 50, 5774–5777 RSC. For the template solvent effects and pillar[n]arenes interconversions see also: (b) T. Ogoshi, N. Ueshima, F. Sakakibara, T. Yamagishi and T. Haino, Org. Lett., 2014, 16, 2896–2899 CrossRef CAS PubMed.
- J. Cao, Y. Shang, B. Qi, X. Sun, L. Zhang, H. Liu, H. Zhang and X. Zhou, RSC Adv., 2015, 5, 9993–9996 RSC.
- (a) Z. B. Zhang, B. Y. Xia, C. Y. Han, Y. H. Yu and F. H. Huang, Org. Lett., 2010, 12, 3285–3287 CrossRef CAS PubMed; (b) T. Ogoshi, T. Aoki, K. Kitajima, S. Fujinami, T. Yamagishi and Y. Nakamoto, J. Org. Chem., 2011, 76, 328–331 CrossRef CAS PubMed; (c) T. Boinski and A. Szumna, Tetrahedron, 2012, 68, 9419–9422 CrossRef CAS.
- (a) M. Holler, N. Allenbach, J. Sonet and J.-F. Nierengarten, Chem. Commun., 2012, 48, 2576–2578 RSC; (b) K. Wang, L.-L. Tan, D.-X. Chen, N. Song, G. Xi, S. X.-A. Zhang, C. Li and Y.-W. Yang, Org. Biomol. Chem., 2012, 10, 9405–9409 RSC.
- Y. Yao, J. Li, J. Dai, X. Chia and M. Xue, RSC Adv., 2014, 4, 9039–9043 RSC.
- O. Branytska and R. Neumann, Synlett, 2004, 1575–1576 CAS.
- For the aliphatic nitriles incapsulation by pillar[6]arenes see: T. Ogoshi, T. Akustu, D. Yamafuji, T. Aoki, K. Kitajima, S. Fujinami, T. Yamagishi and Y. Nakamoto, Angew. Chem., Int. Ed., 2013, 52, 8111–8115 CrossRef CAS PubMed.
- The solvation will affect the solute electronic structure. And the difference will depend on the strength of the solute–solvent interactions. See: (a) C. J. Cramer, Essentials of Computational Chemistry, John Wiley & Sons Ltd., 2004, p. 618 Search PubMed; (b) C. Reichardt and T. Welton, Solvents and Solvent Effects in Organic Chemistry, John Wiley & Sons Ltd., 2011 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental data and NMR spectra for all compounds. See DOI: 10.1039/c5ra19569a |
| ‡ Representative synthetic procedure of pillar[6]arenes 2: to a solution of the appropriate 1,4-dialkoxy benzene (2 mmol) in 2 mL of acetonitrile, paraformaldehyde (124 mg, 4 mmol) was added. The reaction mixture was stirred for 5 minutes. Then conc. H2SO4 (32 μL, 30 mol%) was added to that reaction mixture and stirred at room temperature for 5 minutes. After that 5 mL of ethanol was poured into the reaction mixture. The resulting precipitate was filtered off, washed with ethanol, dried and, depending on the type of pillar[6]arene purified by column chromatography to result in analytically pure peralkylated pillar[6]arenes. Experimental details are provided in the ESI. |
| This journal is © The Royal Society of Chemistry 2015 |