
Substituted furopyridinediones as novel inhibitors of α-glucosidase†
Abstract
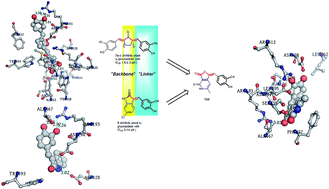
The global preponderance of diabetes mellitus has prompted the medical community to opt for various therapeutic solutions to curb this menace. One of the means involved controlling the post prandial hyperglycemia. α-Glucosidase inhibitors are known to be excellent agents for controlling postprandial hyperglycemia. Different classes of α-glucosidase inhibitors have been discovered. In this context, a diverse library of substituted furopyridinediones (12a–v) was designed as potential inhibitors of α-glucosidase, using an intuitive scaffold hopping approach (which was further rationalized by molecular docking). They were synthesized in one step via an aldol condensation reaction of the furopyridinedione scaffold and appropriate aldehydes. The compounds were screened against α-glucosidase using acarbose as the reference inhibitor. Among the screened compounds, 12p transpired to be the lead candidate with an IC50 of 0.24 μM. Lineweaver–Burke analysis of 12p indicated that it is a mixed inhibitor. X-ray crystallography of 12p further confirmed its structure. Molecular modelling studies and molecular dynamic simulation experiments were performed against a homology model of α-glucosidase to observe the binding interaction of 12p with the enzymes.
 Please wait while we load your content...
Please wait while we load your content...

