
Catalytic properties of γ-Al2O3 supported Pt–FeOx catalysts for complete oxidation of formaldehyde at ambient temperature
Abstract
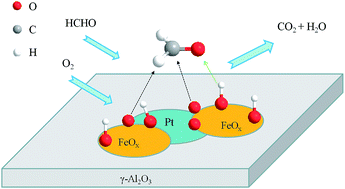
A series of γ-Al2O3 supported Pt–FeOx catalysts (Pt–FeOx/Al2O3) with different Fe/Pt atom ratios were prepared, and their catalytic properties were investigated in the oxidation of formaldehyde. It was found that the catalytic activities of Pt–FeOx/Al2O3 catalysts are varied with the change of Fe/Pt ratios. Among them, the sample with a Fe/Pt ratio of 1.0 exhibits the highest activity, which can efficiently convert formaldehyde to CO2 at ambient temperature. The catalytic activity of the Pt–FeOx/Al2O3 catalyst can be further improved by the addition of water vapor into the feed stream. A variety of characterization results showed that both Pt nanoparticles and FeOx species are highly dispersed on the surface of the γ-Al2O3 support. Changing Fe/Pt ratios could influence the chemical states and the redox properties of Pt and Fe species. The catalysts with appropriate Fe/Pt ratios have more accessible active sites, i.e., the Pt–O–Fe species, which are located at the boundaries between FeOx and Pt nanoparticles, thus showing high activity for the oxidation of formaldehyde under ambient conditions.
 Please wait while we load your content...
Please wait while we load your content...

