
Functionalized boron nitride porous solids†
P. M. Sudeep‡
a,
S. Vinod‡b,
S. Ozdenb,
R. Sruthib,
Akos Kukoveczcd,
Zoltan Konyace,
Robert Vajtaib,
M. R. Anantharamanf,
P. M. Ajayan*b and
Tharangattu N. Narayanan*a
aTIFR-Centre for Interdisciplinary Sciences, Tata Institute of Fundamental Research, Hyderabad-500075, India. E-mail: tnn@tifrh.res.in; tn_narayanan@yahoo.com
bMaterials Science and NanoEngineering Department, Rice University, Houston, TX, USA. E-mail: ajayan@rice.edu
cDepartment of Applied and Environmental Chemistry, University of Szeged, H-6720 Szeged, Rerrich Bela ter 1, Hungary
dMTA-SZTE “Lendület” Porous Nanocomposites Research Group, University of Szeged, H-6720 Szeged, Rerrich Bela ter 1, Hungary
eMTA-SZTE Reaction Kinetics and Surface Chemistry Research Group, University of Szeged, H-6720 Szeged, Rerrich Bela ter 1, Hungary
fDepartment of Physics, Cochin University of Science and Technology, Kochi-682022, Kerala, India
First published on 16th October 2015
Abstract
Hexagonal boron nitride (h-BN), also known as white graphene, is well known for its chemical inertness. Recent studies indicate that functionalization of h-BN can tune its physico-chemical properties, including its electrical conductivity. Here we propose a method for the functionalization of h-BN flakes with various oxygen functionalities to make a graphite oxide analogue of h-BN, with a view to develop cross-linked, low-density (∼40 mg cm−3), and porous h-BN solids, as have been recently well cited for graphene and graphite oxide. For the first time, a macro-porous low density h-BN monolith foam is developed via a single step template free chemical route followed by a lyophilisation process. h-BN is known for its high thermal stability, and here oil adsorption by the foam (∼2 g g−1) and complete burning of the adsorbed oil without disrupting the h-BN skeleton were demonstrated indicating the flexibility of tuning the morphology of the h-BN in bulk, like graphite, without losing its inherent physical properties, opening new avenues for h-BN in the energy and environment related fields.
Ever since the discovery of graphene, research on other 2-dimensional (2D) atomic layers has been always on the increase.1 A number of two dimensional layered materials based on dichalcogenides, perovskites, transition metal oxides and hexagonal boron nitride (h-BN) have garnered immense popularity due to their exciting thermal, mechanical and electronic properties.2–4 Hexagonal boron nitride (h-BN), a wide bandgap layered insulator, known for its chemical inertness and thermal stability,5 is receiving tremendous attention for its applicability in electronics and thermal management.6 Though an electrically insulating material,7 h-BN has been identified as the best ever known substrate for high carrier mobility graphene,8 and is widely used for the bandgap engineering of graphene.9 h-BN is also an ideal candidate for optical components, super hydrophobic surface coatings, substrates for electronic devices, mechanical composites and catalysis.10–13 Though pristine h-BN is an insulator, upon fluorination it is found to become conductive, and this enables functionalized h-BN to act as a potential platform for electronic devices.14
Chemical functionalization of h-BN is a challenge due to its high chemical stability. It was reported that water assisted exfoliated h-BN flakes contain a small amount of oxygen functional groups.15 But studies show that the extent of functionalization is negligible so efficient cross-linking of individual sheets using external cross-linking agents, as we reported for graphene oxide,16–18 is almost impossible. Other non-covalent functionalization approaches include the functionalization of h-BN nanostructures with amine, phosphine and polymers.19–21 The resulting linkages are attributed to π–π interactions and the interactions between boron and nitrogen. Coleman et al. proposed a double step method for the covalent functionalization of exfoliated h-BN nanosheets.22 But the development of a low density, h-BN porous solid by a large scale, mutual linkage of functionalized h-BN sheets is not yet realized, and such a porous solid, if developed, will be useful in many applications including as a solid thermal management material, as a spacer in 3D batteries, for selective organic absorption from water etc.
Here, a method for the functionalization of h-BN flakes is proposed and a covalent linkage between the flakes is attempted using an external cross-linking agent, glutaraldehyde (GAD). The developed porous monoliths were further studied for their structural and morphological properties along with their applicability for oil absorption.
Results and discussion
A method similar to the ‘Improved method’, for the development of graphene oxide from graphite, was adopted for the functionalization of h-BN23 (ESI†). Commercial h-BN (1 μm size, 98% pure, Sigma Aldrich) was used as the starting material. The h-BN foam was synthesized through the chemical cross-linking of functionalized individual h-BN flakes. The functionalized h-BN (i-hBN) was dispersed in deionized water (5 mg mL−1) and treated with resorcinol (11 mM) and GAD (22 mM). The resulting viscous fluid like material was sonicated (135 W, 28 °C) for 3 hours. This slurry was then subjected to lyophilization for 24 hours which resulted in a white low density (bulk density ∼ 40 mg cm−3) h-BN solid as shown in Fig. 1A. | ||
| Fig. 1 (A) Photograph of the h-BN foam. The size of the foam is shown via the adjacent scale. (B) and (C) are the FESEM images of the same h-BN foam showing the microscopic structure. | ||
The XRD pattern of the h-BN foam (ESI S1†) resembles that of bulk h-BN. The porosity of the h-BN foam is evident from the Field Emission Scanning Electron Microscope (FESEM) images shown in Fig. 1B and C. The SEM images indicate the interconnected porous nature of the white solid with “macropore” (pore size > 50 nm) structures.
Further, a Transmission Electron Microscope (TEM) image of the h-BN foam taken after dispersing it in acetone using a mild sonication is shown in Fig. 2A. The presence of a many layered disordered structure of h-BN flakes is evident from the image. This indicates that the ‘Improved method’ could not completely exfoliate h-BN unlike in the case of graphite oxide. In addition, a thermo-gravimetric (TG) analysis (Fig. 2C) was carried out in air on h-BN and i-hBN. It is clear from the analysis that pristine h-BN is stable in air up to ∼750 °C but chemically modified h-BN (i-hBN) starts to disintegrate above 230 °C, which is attributed to the removal of oxygen containing groups (∼25% removal by 750 °C). The TGA study on the h-BN foam is shown in Fig. 2C (bottom). Here, the functional moieties start disintegrating from low temperatures (below 100 °C) onwards, which could be due to the surface adsorbed water molecules in the highly functionalized and porous h-BN foam, and continue to do so up to a temperature of 450 °C (40% removal). The presence of other oxygen containing groups (which is further evident from the FTIR spectra, Fig. 3) induced by the cross-linking process also contribute to the decomposition in this temperature window.
 | ||
| Fig. 2 (A) TEM and (B) HRTEM images of the h-BN foam. (C) TGA of pristine h-BN, i-hBN and h-BN foam. | ||
 | ||
| Fig. 3 FTIR spectra of h-BN, i-hBN and h-BN foam, comparing the functionalities of h-BN foam with pure h-BN powder and functionalised h-BN. | ||
To further probe the chemical functionalization, a Fourier Transform Infrared Spectroscopy (FTIR) study was conducted, and the spectra of h-BN, i-hBN and h-BN foam are compared in Fig. 3. The presence of oxygen bearing functional groups is clear from the FTIR spectra of i-hBN and h-BN foam. Other than the characteristic peaks of B–N, the spectrum of the h-BN foam contains peaks corresponding to B–N–O at 1165 cm−1 and 959 cm−1. The peak at 1100 cm−1 corresponds to the B–O–H in plane bending. It was reported that in the FTIR spectrum of h-BN, it is difficult to find the functional peaks because of the prominent highly intense B–N peaks at 817 cm−1 and 1370 cm−1 which can overlap with other vibrations.24 However in the present case of the h-BN foam, the functional peaks are clear and distinct. The presence of an additional functional group at ∼2900 cm−1 corresponding to –CH stretching indicates the hemiacetal linkage in the h-BN foam as previously reported in the case of GO foam.17 A considerable red shift in the –OH stretching frequency in the h-BN foam compared to i-hBN (i h-BN) also indicates covalent linkage via hemiacetal formation. This establishes the covalent linkages between individual h-BN flakes via hemiacetal linkage due to the hydroxyl groups in the h-BN flakes and the aldehyde groups in the cross-linking agent GAD, resulting in a porous solid.
In addition, the deconvoluted XPS slow scan spectra of B 1s, N 1s, O 1s and C 1s unravel (Fig. 4) the chemical structure of the h-BN foam. Using CASA XPS software, the atomic ratios of B, C, N and O were estimated as 1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.11:1:0.08 respectively in the case of i-hBN, and as 1.2:0.45:1:0.23 in the h-BN foam. The small amount of carbon content is from the amorphous carbon network formation by the resorcinol-GAD cross-linking. Moreover, the shoulder peaks in B, C, N and O are associated with B–O, B–C, N–O, B–N and C–O bonding in agreement with the FTIR analysis. The survey scan XPS spectra of pure h-BN and h-BN foam are shown in Fig. S2 (ESI†). The absence of oxygen in h-BN (pristine) is evident from the survey scan, while the h-BN foam XPS spectrum showed a considerable amount of oxygen, indicating the oxygen functionalization of h-BN after treatment.
:0.11:1:0.08 respectively in the case of i-hBN, and as 1.2:0.45:1:0.23 in the h-BN foam. The small amount of carbon content is from the amorphous carbon network formation by the resorcinol-GAD cross-linking. Moreover, the shoulder peaks in B, C, N and O are associated with B–O, B–C, N–O, B–N and C–O bonding in agreement with the FTIR analysis. The survey scan XPS spectra of pure h-BN and h-BN foam are shown in Fig. S2 (ESI†). The absence of oxygen in h-BN (pristine) is evident from the survey scan, while the h-BN foam XPS spectrum showed a considerable amount of oxygen, indicating the oxygen functionalization of h-BN after treatment.
 | ||
| Fig. 4 Deconvoluted XPS spectra of 3D h-BN foam: (A) B 1s (B) N 1s (C) C 1s and (D) O 1s. | ||
To further investigate the hydrophobicity of h-BN, a contact angle measurement was carried out and is exhibited in Fig. S3 (ESI†). Pristine h-BN (commercial) shows hydrophobicity with a contact angle of 130°, as reported in the literature.25 But in the case of the h-BN foam, water is wetting the surface, which indicates the increased amount of oxygen functionalities at the surface.
Nitrogen adsorption–desorption isotherms (Fig. 5) of the h-BN foam were measured using a Quantachrome NOVA 3000 instrument at 77 K. The h-BN foam was outgassed at 200 °C for 2 hours under dynamic vacuum before each measurement.
 | ||
| Fig. 5 The adsorption–desorption isotherm pairs of the h-BN foam. | ||
The h-BN foam exhibited an IUPAC type II isotherm which is characteristic of either non-porous or macroporous adsorbents. The isotherms are linear at very low relative pressure up to the inflection point, Prel > 0.9 where N2 condensation finally occurs. This is a macroporous structure built up of flat lamellae separated by several hundreds of nanometers. The layers forming these macropore walls are non-porous themselves. A macro-world analogue of this structure would be a house of cards.
One of the advantages of h-BN is its high thermal stability in air. These macro-porous monoliths were demonstrated for oil absorption (Fig. 6). The complete soaking of the foam in oil resulted in the holding of 2 g g−1 oil (2 g used engine oil in 1 g foam, this value is similar to the commercial polymer beds used in an oil recovery field26). After careful removal of the un-adsorbed surface oil, burning of the adsorbed oil in air was conducted as shown in Fig. 6. The FTIR spectrum of the residual h-BN foam after the complete burning of the oil is shown in Fig. 7. It is observed that even after the burning of the soaked oil, the h-BN foam still has functional groups such as –OH and –CH stretching bonds along with the characteristic BN bonds as discussed in the previous section indicating the structural integrity of the developed h-BN foam.
 | ||
| Fig. 6 Schematic of the h-BN foam development and its oil adsorption and burning. Real time images of the oil (used engine oil) soaking and later burning of the foam are shown on the right. | ||
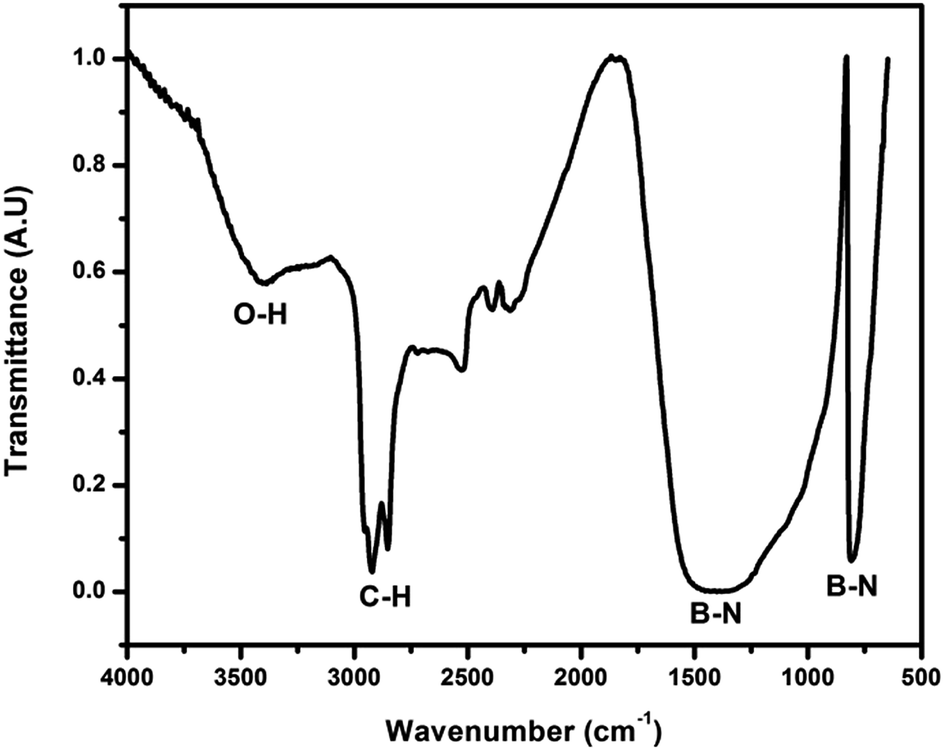 | ||
| Fig. 7 FTIR spectrum of the h-BN foam after the burning of absorbed oil. | ||
In conclusion, a method for the development of functionalized h-BN was pursued and oxygenated h-BN flakes were developed in bulk. These h-BN flakes were covalently bonded together using GAD-resorcinol cross-linking chemistry to obtain a low density macro-porous h-BN monolith solid (foam). The structural integrity, chemical functionalization and chemical structure of the h-BN foams were studied using XRD, FESEM, FTIR and XPS analyses. This functionalization approach and template free bulk method for the synthesis of h-BN porous solids open new avenues for h-BN in applications such as corrosion resistive stable coatings, filter membranes, thermal foam, selective oil adsorption beds and spacers in 3-dimensional batteries. One such application is demonstrated here showing the capability of holding organic contaminants in the porous h-BN and their subsequent burning without oxidizing or disrupting the h-BN skeleton.
Acknowledgements
P. M. S. and T. N. N. acknowledge the financial support from TIFR-Centre for Interdisciplinary Sciences. TNN acknowledges DST for financial support in the form of DST-FAST Track scheme (SB/FTP/PS-084/2013). P. M. S., S. V., R. S., R. V. and P. M. A. acknowledge the U.S. Department of Defense: U.S. Air Force Office of Scientific Research for the Project MURI: “Synthesis and Characterization of 3-D Carbon Nanotube Solid Networks” Award No. FA9550-12-1-0035. A. K. and Z. K. acknowledge the financial support of the OTKA NN 110676 and K 112531 projects.References
- A. Nag, K. Raindogia, K. P. S. S. Hembram, R. Datta, U. V. Wangmare and C. N. R. Rao, ACS Nano, 2010, 4, 1539–1544 CrossRef CAS PubMed.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS PubMed.
- B. Radisavljevic, A. Radenovic, J. Brivio, V. Giacometti and A. Kis, Nat. Nanotechnol., 2011, 6, 147–150 CrossRef CAS.
- C. R. Dean, A. F. Young, I. Meric, C. Lee, L. Wang, S. Sorgenfrei, K. Watanabe, T. Taniguchi, P. Kim, K. L. Shepard and J. Hone, Nat. Nanotechnol., 2010, 5, 722–726 CrossRef CAS PubMed.
- R. Arenal, X. Blase and A. Loiseau, Adv. Phys., 2010, 59, 101–179 CrossRef CAS.
- W.-Q. Han, Anisotropic Hexagonal Boron-Nitride Nanomaterials: Synthesis and Applications, Nanotechnologies for the Life Sciences, Wiley, New York, published online Oct. 15, 2010 Search PubMed.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De and I. T. McGovern, et al., Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- J. N. Coleman, et al., Science, 2011, 331, 568–571 CrossRef CAS PubMed.
- U. Khan, A. O’Neill, M. Lotya, S. De and J. N. Coleman, Small, 2010, 6(7), 864–871 CrossRef CAS PubMed.
- D. Golberg, Y. Bando, Y. Huang, T. Terao, M. Mitome, C. Tang and C. Zhi, ACS Nano, 2010, 4, 2979–2993 CrossRef CAS PubMed.
- A. Pakdel, C. Zhi, Y. Bando, T. Nakayama and D. Golberg, ACS Nano, 2011, 5, 6507–6515 CrossRef CAS PubMed.
- J. Yu, L. Qin, Y. Hao, S. Kuang, X. Bai, Y. M. Chong, W. Zhang and E. Wang, ACS Nano, 2010, 4, 414–422 CrossRef CAS PubMed.
- J. C. S. Wu, Z. A. Lin, J. W. Pan and M. H. Rei, Appl. Catal., A, 2001, 219, 117–124 CrossRef CAS.
- Y. Xue, Q. Liu, G. He, K. Xu, L. Jiang, X. Hu and J. Hu, Nanoscale Res. Lett., 2013, 8, 49 CrossRef PubMed.
- Y. Lin, T. V. Williams, T. B. Xu, W. Cao, H. E. Elsayed-Ali and J. W. Connell, J. Phys. Chem. C, 2011, 115, 2679–2685 CAS.
- S. Vinod, C. S. Tiwary, P. A. S. Autreto, J. Taha-Tijerina, S. Ozden, A. C. Chipara, R. Vajtai, D. S. Galvao, T. N. Narayanan and P. M. Ajayan, Nat. Commun., 2014, 5, 4541 CAS.
- P. M. Sudeep, T. N. Narayanan, A. Ganesan, M. M. Shaijumon, H. Yang and S. Ozden, et al., ACS Nano, 2013, 7(8), 7034–7040 CrossRef CAS PubMed.
- M. R. Philip, T. N. Narayanan, S. Bhushan Arya and D. K. Pattanayak, J. Mater. Chem. A, 2014, 2, 19488–19494 CAS.
- W. Wang, Y. Bando, C. Zhi, W. Fu, E. Wang and D. Golberg, J. Am. Chem. Soc., 2008, 130, 8144–8145 CrossRef CAS.
- C. Zhi, Y. Bando, C. Tang and D. Golberg, J. Am. Chem. Soc., 2005, 127, 17144–17145 CrossRef CAS PubMed.
- Y. Lin, T. V. Williams and J. W. Connell, J. Phys. Chem. Lett., 2010, 1, 277–283 CrossRef CAS.
- T. Sainsbury, A. Satti, P. May, Z. Wang, I. McGovern, Y. K. Gun’ko and J. Coleman, J. Am. Chem. Soc., 2012, 134, 18758–18771 CrossRef CAS PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun and A. Slesarev, et al., ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- A. S. Nazarov, V. N. Demin, E. D. Grayfer, A. I. Bulavchenko, A. T. Arymbaeva, H. Shin, J.-Y. Choi and V. E. Fedorov, Chem.–Asian J., 2012, 7, 554–560 CrossRef CAS PubMed.
- E. Hussain, T. N. Narayanan, J.-T. Tijerena, S. Vinod, R. Vajtai and P. M. Ajayan, ACS Appl. Mater. Interfaces, 2013, 5(10), 4129–4135 Search PubMed.
- P. Thanikaivelan, T. N. Narayanan, B. K. Pradhan and P. M. Ajayan, Sci. Rep., 2012, 2, 1–7 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra19091f |
| ‡ These authors are equally contributed. |
| This journal is © The Royal Society of Chemistry 2015 |