DOI:
10.1039/C5RA19062B
(Paper)
RSC Adv., 2015,
5, 93607-93614
A new design strategy on cage insensitive high explosives: symmetrically replacing carbon atoms by nitrogen atoms followed by the introduction of N-oxides
Received
16th September 2015
, Accepted 26th October 2015
First published on 28th October 2015
Abstract
In this work, using hexaprismane as a base skeleton, we designed a novel cage energetic compound 1,3,5,7,9,11-hexaazahexaprismane-1,3,5,7,9,11-hexaoxdies (HAHHO) by employing a new design strategy: symmetrically replacing six carbon atoms by nitrogen atoms in hexaprismane followed by the introduction of six N-oxides. Its detonation performance and sensitivity were estimated by using the density functional theory method. It was found that HAHHO possesses much higher energetic performance than 1,3,5,7-tetranitro-1,3,5,7-tetrazocane and lower sensitivity than 2,4,6-trinitrotoluene, suggesting that its overall performance are outstanding and may be considered as the potential candidate of insensitive high explosives. The special double cage structure of HAHHO may be an important reason why it has low sensitivity. The results show that our strategy used for designing HAHHO is practical and may be applied to design and develop other cage explosives with high energy and low sensitivity.
1 Introduction
In the past several decades, to meet the rapid development of modern industry, many studies1–15 have been done on finding and synthesizing ideal insensitive high explosives (IHE) coupled with the low sensitivity of the widely used insensitive explosive TNT (2,4,6-trinitrotoluene) and the high energy of the commonly used high explosive HMX (1,3,5,7-tetranitro-1,3,5,7-tetrazocane). However, though a lot of human and material resources were devoted and a great many of new energetic compounds were synthesized by introducing different many energetic substituent groups into different carbocycle and N-heterocycle skeleton mainly, few of them achieved this standard. One important reason for this is that the energy of the basic ring skeleton carbocycle and N-heterocycle are low generally, thus, many energetic substituent groups are needed to improve the detonation performance, which would increase the sensitivity dramatically in the meantime. If decrease the amounts of energetic substituent groups, the energy properties of the compounds would be mediocre. The balance between the energy and sensitivity is still an unsolved big problem, much more studies are inquired to obtain new IHE with comparative sensitivity and energy to TNT and HMX, respectively.
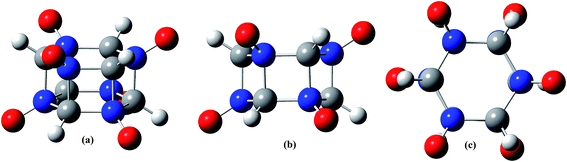
Lately, cage explosives become a research hot in energetic materials field because of the outstanding detonation performance. Different to those of the carbocycle and N-heterocycle skeleton, the cage skeleton contains very high energy. For example, the cage cubane16,17 and hexaprismane18,19 (Fig. 1), their heats of formation (622 kJ mol−1 for cubane and 680 kJ mol−1 for hexaprismane) are both very high. Based on them, two cage explosives octanitrocubane (ONC)20 and dodecanitrohexaprismane21 (DNH, Fig. 1) can be obtained by replacing all hydrogen atoms by nitro groups (Fig. 1a). ONC and DNH both have super high energy, and their detonation performance are higher than all of the synthesized carbocyclic and N-heterocyclic energetic compounds till now. However, due to the too many nitro groups in the structure, both of them are not very insensitive and difficult to synthesize, especially for DNH, which is estimated to be as sensitive as HMX and have not been synthesized successfully till now. Thus, one strategy (Fig. 1b) was used to decrease the amount of nitro groups and the sensitivity without reducing the energy obviously: first, half the carbon atoms in the cage skeleton are replaced by using nitrogen atoms symmetrically to form an aza-cage skeleton, then, all the hydrogen atoms in the aza-cage skeleton are substituted by nitro groups. Through this method, half of nitro groups are removed and the sensitivity is reduced significantly without decreasing the energy dramatically.21 This means that a better balance between the energy and sensitivity were achieved. However, despite this, the estimated sensitivity of the resulted compound hexanitrohexaazaprismane (HNHAH, Fig. 1) is still obviously higher than that of TNT, though its energy is much higher than HMX. Therefore, other improved strategies are needed.
 |
| | Fig. 1 Molecular frameworks of HAHHO. | |
In the present study, based on hexaprismane, a novel cage energetic compound 1,3,5,7,9,11-hexaazahexaprismane-1,3,5,7,9,11-hexaoxdies (HAHHO, Fig. 1 and 2) was designed by employing a new design strategy (Fig. 1c): first, symmetrically replacing six carbon atoms by nitrogen atoms in hexaprismane to form 1,3,5,7,9,11-hexaazahexaprismane (HAH), the density and heat of formation (HOF) would be improved obviously by this N hybridization. Then, symmetrically introducing six N-oxides into HAH to generate HAHHO, the density, HOF and oxygen balance (OB) can be enhanced significantly through this step. It can be expected that HAHHO has extremely high HOF and superior density, which will keep its energy in a higher level. Because that there are no nitro groups or weak and sensitive N–N bonds in HAHHO, it is not likely possesses high sensitivity. In addition, eighteen intramolecular hydrogen bonds (Fig. 6) may be formed between the six oxygen atoms and six hydrogen atoms, which would reduce its sensitivity to some degree. These indicate that HAHHO is expected to be with high energy and low sensitivity, this inference will be verified by using density functional theory (DFT) in the following section.
 |
| | Fig. 2 (a) The optimized structure of HAHHO. (b) and (c) The perspective view of HAHHO from other viewpoints. White, red, blue, and gray spheres stand for H, O, N, and C atoms, respectively. | |
2 Computational methods
The calculations of gas-phase heats of formation of HAHHO was carried out for the atomization reaction CaHbOcNd → aC (g) + bH (g) + cO (g) + dN (g) by using the CBS-4M theory.
According to Hess's law of constant heat summation,22 the solid-phase heat of formation can be obtained from the gas-phase heat of formation (ΔHf,gas) and heat of sublimation (ΔHsub):
| | |
ΔHf,solid = ΔHf,gas − ΔHsub
| (1) |
Politzer et al.23,24 reported that the heat of sublimation correlates with the molecular surface area and the electrostatic interaction index νσtot2 for energetic compounds. The empirical expression of the approach is as follows:
| | |
ΔHsub = aA2 + b(νσtot2)0.5 + c
| (2) |
where
A is the surface area of the 0.001 electrons per bohr
3 isosurface of the electronic density of the molecule,
ν describes the extent of balance between positive potential and negative potential on the isosurface, and
σtot2 is a measure of the variability of the electrostatic potential on the molecular surface. The coefficients
a,
b, and
c have been determined by Rice
et al.:
a = 2.670 × 10
−4 kcal mol
−1 A
4,
b = 1.650 kcal mol
−1, and
c = 2.966 kcal mol
−1.
25 The descriptors
A,
ν, and
σtot2 were calculated by using the computational procedures proposed by Bulat
et al.26 This approach has been demonstrated to predict reliably the heats of sublimation of many energetic organic compounds.
13–15 These calculations were carried out at the B3LYP/6-311++G(2df,2p)//B3LYP/6-31G(d).
24
The infrared (IR) and ultraviolet-visible (UV-vis) spectrums were calculated by the B3LYP/6-31+G(d, p) method.
The detonation velocity and pressure were estimated by the Kamlet–Jacobs equations27 as
| |
D = 1.01(N![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) 1/2 Q1/2)1/2(1 + 1.30ρ) 1/2 Q1/2)1/2(1 + 1.30ρ)
| (3) |
| | |
P = 1.558ρ2N1/2Q1/2
| (4) |
where each term in the equations of
(1) and
(2) is defined as follows:
D, the detonation velocity (km s
−1);
P, the detonation pressure (GPa);
N, the moles of detonation gases per gram explosive;
, the average molecular weight of these gases;
Q, the heat of detonation (cal g
−1); and
ρ, the loaded density of explosives (g cm
−3). For known explosives, their
Q and
ρ can be measured experimentally; thus their
D and
P can be calculated according to
eqn (1) and
(2). However, for some compounds, their
Q and
ρ cannot be evaluated from experimental measures. Therefore, to estimate their
D and
P, we first need to calculate their
Q and
ρ. The detonation products are supposed to be only CO
2, H
2O, and N
2, so released energy in the decomposition reaction reaches its maximum.
The theoretical density was obtained using an improved equation proposed by Politzer et al.28 in which the interaction index νσtot2 was introduced:
| |
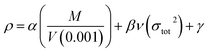 | (5) |
where
M is the molecular mass (g mol
−1) and
V(0.001) is the volume of the 0.001 electrons per bohr
3 contour of electronic density of the molecule (cm
3 per molecule). The coefficients
α,
β, and
γ are 0.9183, 0.0028, and 0.0443, respectively. These calculations were carried out at the density functional B3PW91/6-31G(d, p) level.
28
The strength of bonding, which could be evaluated by bond dissociation energy (BDE), is fundamental to understand chemical processes.29 The energy required for bond homolysis at 298 K and 1 atm corresponds to the enthalpy of reaction A − B (g) → A˙(g) + B˙(g), which is the bond dissociation enthalpy of the molecule A–B by definition.30 For many organic molecules, the terms “bond dissociation energy” and “bond dissociation enthalpy” usually appear interchangeably in the literature.31 Thus, at 0 K, the homolytic bond dissociation energy can be given in terms of eqn (6):
| | |
BDE0 (A–B) = E0 (A˙) + E0 (B˙) − E0 (A–B)
| (6) |
The bond dissociation energy with zero-point energy (ZPE) correction can be calculated by eqn (7):
| | |
BDE (A–B)ZPE = BDE0 (A–B) + ΔEZPE
| (7) |
where Δ
EZPE is the difference between the ZPEs of the products and the reactants.
The free space per molecule in the unit cell, designated ΔV, can be used to estimate the impact sensitivity of an energetic compound.32 ΔV can be represented as the difference between the effective volume per molecule that would be required to completely fill the unit cell, Veff, and the intrinsic gas phase molecular volume, V (0.003):
| | |
ΔV = Veff − Vint = M/ρ − V(0.003)
| (8) |
where
V(0.003) is defined as the volume enclosed by the 0.003 electrons per bohr
3 contour of the molecule's electronic density.
M is the molecular mass and
ρ is the crystal density. These calculations were carried out at the density functional B3PW91/6-31G(d, p) level.
32
Other calculations were performed at the B3LYP/6-31+G(d, p) level with the Gaussian 03 package.33 In the geometry optimization, the maximum force was converged less than 0.00045 eV Å−1, the RMS force less than 0.0003 eV Å−1, the maximum displacement less than 0.0018 Å, and the RMS displacement less than 0.0012 Å. All of the optimized structures were characterized to be true local energy minima on the potential energy surfaces without imaginary frequencies.
Since high energy explosives are in condensed phases usually, especially in solid forms, we predicted the crystal structure of HAHHO by searching the possible molecular packing among ten probable space groups (P21/c, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , P212121, Pbca, C2/c, P21, Pna21, C2, CC, and Pbcn).34–36
, P212121, Pbca, C2/c, P21, Pna21, C2, CC, and Pbcn).34–36
3 Results and discussion
3.1 HOF and energetic properties
Table 1 displays the calculated solid-phase HOF, ρ, Q, D, and P of HAHHO and ONC. First, it is found that the calculated values of ONC in this work are very close to the experimental results or previous calculated results. Then, it is seen that the HOF of HAHHO is much higher than that of ONC, and the HOF of HAHHO is even comparable with that of one nitrogen-high compound 1,1′-azobis(tetrazole) (6.2 kJ g−1)38 which has extremely high heat of formation. This indicates that the HOF property of cage HAHHO is very outstanding and this is mainly derived from its original cage skeleton that contains very high energy. The high HOF of HAHHO makes it possesses extremely high Q, which is also significantly higher than that of ONC, this further makes HAHHO has comparative D and P with ONC, though the ρ of former is obviously lower than that of the later. In a word, though there are no nitro groups or any other energetic substituent groups, the detonation performance of HAHHO is comparable with ONC. Fig. 3 displays a comparison of Q, D, and P of HMX, CL-20 (2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane), ONC, HAHHO, HNHAH and DNH. First, HAHHO has the highest Q. Then, the D HAHHO of is only lower than that of DNH but is higher than those of the rest. Finally, HAHHO has lower P than DNH and CL-20, and its P is close to ONC and HNHAH and higher than HMX. In all, the energetic properties of HAHHO is obviously higher than that of HMX and comparable with those of ONC and CL-20, while these two explosives are the two most powerful high explosives composed of C, H, O, and N that have been synthesized until now. This means that the detonation performance of HAHHO is very remarkable, though there are no nitro groups or any other energetic substituent groups in its structure. Thus, the energy goal of finding new IHE with comparative sensitivity and energy to TNT and HMX, respectively, has been achieved.
Table 1 Solid-phase HOF (kJ g−1), densities (ρ, g cm−3), Q (kJ g−1), D (km s−1), and P (GPa) of HAHHO and ONC
| Compound |
HOF |
ρ |
Q |
D |
P |
| Experimental values from ref. 1 and 37, respectively. Calculated values from ref. 21. |
| ONC |
1.8 (1.8a) |
1.97 (1.97b, 1.98a) |
8.2 (8.2b) |
9.6 (9.6b) |
43.6 (43.5b) |
| HAHHO |
6.0 |
1.88 |
11.1 |
9.7 |
42.8 |
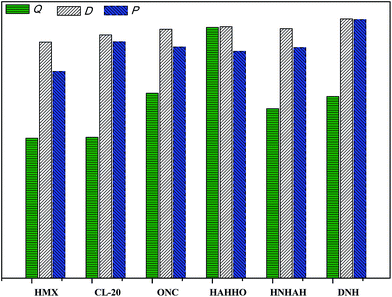 |
| | Fig. 3 A comparison of Q, D, and P of HMX, CL-20, ONC, HAHHO, HNHAH, and DNH. | |
3.2 Thermal stability and sensitivity
For an ideal IHE, both high energy and low sensitivity are required. In this section, we turn to investigate the thermal stability and sensitivity of HAHHO. The BDE can provide useful information for understanding the stability of energetic materials. Generally, the smaller energy for breaking a bond is, the weaker the bond is, and the easier the bond becomes a trigger bond; that is to say, the corresponding compound is more unstable and its sensitivity is larger. However, it should be noted that the bond energies are not always a good measure of thermal stability since there are various possible mechanisms of decomposition while breaking a trigger linkage is only one of them. The natural bond orders of C–N bonds (about 0.78–0.82) in the cage skeleton are obviously lower than those of C–H bonds (about 0.92) and N–O bonds (about 1.2), suggesting that C–N bonds are weaker than C–H bonds and N–O bonds. Thus, we calculated the BDE of C–N bonds. There are two kinds of C–N bonds, the first one is the C–N bond in the hexagon (labeled as C1–N1) while the other one is the C–N in the quadrangle (labeled as C2–N2). The BDE values of C1–N1 bond and C2–N2 bond are calculated to be 222.5 and 126.9 kJ mol−1, showing that HAHHO has good thermal stability and the breaking of C–N bonds in the quadrangle is an initial decomposition step of HAHHO. Fig. 4 gives a comparison of ΔV of DNH, CL-20, ONC, HNHAH, TNT and HAHHO. The free space per molecule in the unit cell, designated ΔV, can be used to estimate the impact sensitivity of an energetic compound.32 Generally, the lower the ΔV value is, the less sensitive the compound is. From Fig. 4, it can be seen that the ΔV value decreases in the order of DNH, CL-20, ONC, HNHAH, TNT and HAHHO, indicating that the sensitivity reduces in the same sequence. This means that HAHHO is more insensitive than other four cage high explosives and is even less sensitive than TNT (Table 2). Thus, HAHHO is a very insensitive explosive, and the sensitivity goal of finding new IHE with comparative sensitivity and energy to TNT and HMX, respectively, has been achieved also. The low sensitivity of HAHHO may be derived from its symmetrical, conjugated and special double cage structure. Fig. 5 displays the HOMO and LUMO of HAHHO, from which it can be seen that almost all atoms all included in the HOMO and LUMO, indicating that this molecule is a big conjugated system. The calculated bond lengths of all C–N, N–O and C–H bonds in HAHHO are found to be close each other, respectively, suggesting that this system has good symmetry in geometry. Fig. 6 displays the hydrogen bonding and electrostatic potential (ESP) of HAHHO. From Fig. 6a–c, it can be seen that eighteen intramolecular hydrogen bonds are formed between the six oxygen atoms and six hydrogen atoms and it looks like that the internal small cage C–N skeleton is surrounded by the external big cage hydrogen bonds. This special double cage structure is obviously different from any other known energetic compounds and may be an important reason why HAHHO is very insensitive though it even has comparable detonation performance with CL-20 and ONC. Previous studies reported that the electrostatic potential (ESP) is related to the impact sensitivity of the energetic material, and the stability can be expressed as a function of the imbalance between positive and negative regions.39–41 In the N–O systems, the regions of stronger positive potential are concentrated on the nitrogen atom and lead to the atypical imbalance which causes the high impact sensitivity. However, it is seen in Fig. 6d that the positive potential is dispersed at the center of the cage skeleton, which may reduce its impact sensitivity effectively.
 |
| | Fig. 4 A comparison of ΔV of DNH, CL-20, ONC, HNHAH, TNT and HAHHO. | |
 |
| | Fig. 5 HOMO and LUMO of HAHHO. | |
 |
| | Fig. 6 Hydrogen bonding (displayed as the dotted lines) of HAHHO (a–c) and ESP (d) [0.001 electron per bohr3 isosurface, color coding: from red (negative) to blue (positive)] of HAHHO. White, red, blue, and gray spheres stand for H, O, N, and C atoms, respectively. | |
Table 2 ΔV values of HAHHO, TNT and CL-20
| Compound |
ΔV (Å3) |
| Calculated values from ref. 29. |
| HAHHO |
51 |
| TNT |
58 (58a) |
| CL-20 |
86 (86a) |
Overall, though with a relatively simple structure and there are no nitro groups and any other energetic substituent groups in the system, HAHHO has comparative detonation performance with CL-20 and ONC, higher energy than HMX, and lower sensitivity than TNT, indicating that its overall performance is outstanding and it may be a very attractive candidate for experiments. Thus, a new potential cage IHE HAHHO coupled with high energy of HMX and low sensitivity of TNT has been obtained successfully, our new strategy used for designing HAHHO is practical and may be applied to design and develop other cage explosives with high energetic properties and low sensitivity.
3.3 Spectral properties
The IR and UV-vis (in dimethylsulfoxide solution) spectrums of HAHHO are displayed in Fig. 7. For the IR spectrum, the strong peaks at 1274, 1218 and 1169 cm−1 is associated with a C–N stretch and N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O symmetric stretch motion. The strong peak at 3220 cm−1 corresponds to the C–H stretch modes. For the UV-vis spectrum, the wide and strong absorption region around at 357 nm correspond n → π* transition of NO bonds and π → π* of the conjugated system.
O symmetric stretch motion. The strong peak at 3220 cm−1 corresponds to the C–H stretch modes. For the UV-vis spectrum, the wide and strong absorption region around at 357 nm correspond n → π* transition of NO bonds and π → π* of the conjugated system.
 |
| | Fig. 7 The calculated IR and UV-vis spectrums of HAHHO. | |
3.4 Predicted crystal properties
In this section, we will predict crystal packing of HAHHO. Dreiding42 is a common force field which is able to produce the condensed-phase properties reliable for a lot of organic systems. Here the Dreiding field was used to predict the crystal structure of HAHHO. The predicted results are presented in Table 3. It is seen that the structure with P symmetry (Fig. 8) has the lowest energy and thus HAHHO most probably belongs to the P space group since the stable polymorph often possesses lower Gibbs free energy of or total energy. It is found that the density (1.90 g cm−3) of HAHHO predicted by the Dreiding force field is close to the calculated value (1.88 g cm−3) in the above section. Thus, the lattice parameters of HAHHO are Z = 4, a = 7.27 Å, b = 14.23 Å, c = 5.22 Å, α = 97.0°, β = 76.2°, and γ = 120.7°. Then, based on the predicted structure with P symmetry, the density of states (DOS) of HAHHO was calculated and displayed in Fig. 9. It can be seen that expected that the C states, N states, O states and H states all make contributions to the valence band and conduction band, indicating that the molecule is a well conjugated system.
Table 3 Unit cell parameters of the possible molecular packing of HAHHO in the ten possible space groups
| Space groups |
P21/c |
P |
P212121 |
Pbca |
C2/c |
P21 |
Pna21 |
C2 |
CC |
Pbcn |
| E in kJ mol−1 cell. |
| Z |
4 |
2 |
4 |
8 |
8 |
2 |
4 |
4 |
4 |
8 |
| Ea |
216.06 |
215.33 |
217.19 |
217.32 |
216.00 |
216.52 |
216.19 |
216.20 |
216.30 |
216.75 |
| ρ (g cm−3) |
1.865 |
1.902 |
1.814 |
1.817 |
1.870 |
1.840 |
1.866 |
1.852 |
1.870 |
1.844 |
| a (Å) |
14.60 |
7.27 |
12.16 |
7.40 |
37.15 |
7.41 |
10.17 |
12.50 |
9.96 |
12.45 |
| b (Å) |
5.21 |
14.23 |
7.69 |
20.59 |
5.19 |
12.43 |
12.31 |
7.36 |
12.31 |
10.42 |
| c (Å) |
14.15 |
5.22 |
10.10 |
12.39 |
14.42 |
5.22 |
7.34 |
10.02 |
10.48 |
14.34 |
| α (°) |
90.0 |
97.0 |
90.0 |
90.0 |
90.0 |
90.0 |
90.0 |
90.0 |
90.0 |
90.0 |
| β (°) |
58.7 |
76.2 |
90.0 |
90.0 |
138.8 |
75.6 |
90.0 |
99.9 |
134.5 |
90.0 |
| γ (°) |
90.0 |
120.7 |
90.0 |
90.0 |
90.0 |
90.0 |
90.0 |
90.0 |
90.0 |
90.0 |
 |
| | Fig. 8 Most possible packing of HAHHO. | |
 |
| | Fig. 9 The density of states (DOS) of HAHHO. | |
4 Conclusions
In this work, we used hexaprismane as a base skeleton to design a novel cage energetic compound HAHHO by employing a new design strategy: symmetrically replacing six carbon atoms by nitrogen atoms in hexaprismane to form HAH, followed by symmetrically introducing six N-oxides into HAH to generate HAHHO. The structure and properties were studied by using the DFT method. The results indicate that HAHHO is a symmetrical and conjugated molecule and most probably belongs to the P space group. Though the structure is simple and no nitro groups or any other energetic substituent groups existed in the structure, HAHHO has comparable detonation performance with ONC and CL-20, its high energy may be its original cage skeleton. HAHHO possesses much higher energetic performance than HMX and lower sensitivity than TNT, suggesting that its overall performance are outstanding and may be considered as the potential candidate of insensitive high explosives. The special double cage structure of HAHHO may be an important reason why it has low sensitivity. In all, a new potential cage IHE, HAHHO, which coupled with high energy of HMX and low sensitivity of TNT, has been obtained successfully, thus, our new strategy used for designing HAHHO is practical and may be applied to design and develop other cage explosives with high energetic properties and low sensitivity.
Acknowledgements
This work was supported by the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (14KJD430002), the Science Foundation of Nanjing Institute of Technology (ZKJ201402, QKJA201202), the Jiangsu Key Laboratory Opening Project of Advanced Structural Materials and Application Technology (ASMA201408), the Youth natural science foundation of Jiangsu Province (BK20130747), and the Science Foundation of Nanjing Institute of Technology (ZKJ201301).
References
- M. X. Zhang, P. E. Eaton, R. Gilardi, N. Gelber, S. Iyel and R. Surapaneni, Propellants, Explos., Pyrotech., 2002, 27, 1–6 CrossRef.
- Y. H. Joo and J. M. Shreeve, Angew. Chem., Int. Ed., 2009, 48, 564–567 CrossRef CAS.
- T. M. Klapötke, C. M. Sabaté and M. Rasp, J. Mater. Chem., 2009, 19, 2240–2252 RSC.
- T. M. Klapötke, F. A. Martin and J. Stierstorfer, Angew. Chem., Int. Ed., 2011, 50, 4227–4229 CrossRef.
- G. H. Tao, Y. Guo, D. A. Parrish and J. M. Shreeve, J. Mater. Chem., 2010, 20, 2999–3005 RSC.
- Q. H. Lin, Y. C. Li, Y. Y. Li, Z. Wang, W. Liu, C. Qi and S. P. Pang, J. Mater. Chem., 2012, 22, 666–674 RSC.
- P. Tin, Q. H. Zhang, J. H. Zhang, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2013, 1, 7500–7510 Search PubMed.
- D. Chand, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2013, 1, 15383–15389 CAS.
- P. Yin, J. H. Zhang, C. L. He, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2014, 2, 3200–3208 CAS.
- J. S. Murray, J. M. Seminario and P. Politzer, Struct. Chem., 1991, 2, 153–166 CrossRef CAS.
- P. Politzer, P. Lane and J. S. Murray, Cent. Eur. J. Energ. Mater., 2013, 10, 37–52 CAS.
- J. J. Zhang, H. C. Du, F. Wang, X. D. Gong and Y. S. Huang, J. Phys. Chem. A, 2011, 115, 6617–6621 CrossRef CAS PubMed.
- F. Wang, H. C. Du, J. H. Zhang and X. D. Gong, J. Phys. Chem. A, 2011, 115, 11788–11795 CrossRef CAS PubMed.
- Q. Wu, Y. Pan, X. L. Xia, Y. L. Shao, W. H. Zhua and H. M. Xiao, Struct. Chem., 2013, 24, 1579–1590 CrossRef CAS.
- Q. Wu, W. H. Zhu and H. M. Xiao, J. Chem. Eng. Data, 2013, 58, 2748–2762 CrossRef CAS.
- B. D. Kybett, S. Carroll, P. Natalis, D. W. Bonnell, J. L. Margrave and J. L. Franklin, J. Am. Chem. Soc., 1966, 88, 626 CrossRef CAS.
- K. A. Lukin, J. C. Li, P. E. Eaton, N. Kanomata, J. Hain, E. Punzalan and R. J. Gilardi, J. Am. Chem. Soc., 1997, 119, 9591–9602 CrossRef CAS.
- G. W. Schriver and D. J. Gerson, J. Am. Chem. Soc., 1990, 112, 4723–4729 CrossRef CAS.
- F. L. Liu, Chin. J. Struct. Chem., 2003, 22, 97–102 CAS.
- A. M. Astakhov, R. S. Stepanov and A. Yu, Babushkin, Combustion, Explosion, and Shock Waves, 1998, vol. 34, pp. 85–87 Search PubMed.
- Q. Wu, W. H. Zhu and H. M. Xiao, RSC Adv., 2014, 4, 3789–3797 RSC.
- P. W. Atkins, Physical chemistry, Oxford University Press, Oxford, 1982 Search PubMed.
- P. Politzer, J. S. Murray, M. E. Grice, M. DeSalvo and E. Miller, Mol. Phys., 1997, 91, 923–928 CrossRef CAS.
- P. Politzer and J. S. Murray, Cent. Eur. J. Energ. Mater., 2011, 8, 209–220 CAS.
- E. F. C. Byrd and B. M. Rice, J. Phys. Chem. A, 2006, 110, 1005–1013 CrossRef CAS PubMed.
- F. A. Bulat, A. Toro-Labbe, T. Brinck, J. S. Murray and P. Politzer, J. Mol. Model., 2010, 16, 1679–1691 CrossRef CAS PubMed.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS.
- P. Politzer, J. Martinez, J. S. Murray, M. C. Concha and A. Toro-Labbé, Mol. Phys., 2009, 107, 2095–2101 CrossRef CAS.
- S. W. Benson, Thermochemical kinetics, Wiley-Interscience, New York, 2nd edn, 1976 Search PubMed.
- I. Mills, T. Cvitas, K. Homann, N. Kallay and K. Kuchitsu, Quantities, units, and symbols in physical chemistry, Blackwell, Oxford, 1988 Search PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- P. Politzer and J. S. Murray, J. Mol. Model., 2014, 20, 2223–2230 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. AlLaham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Pittsburgh, PA, 2003 Search PubMed.
- A. J. Wilson, Acta Crystallogr., Sect. A: Found. Crystallogr., 1988, 44, 715–724 CrossRef.
- R. Srinivasan, Acta Crystallogr., Sect. A: Found. Crystallogr., 1992, 18, 917–918 CrossRef.
- A. D. Mighell, V. L. Himes and J. R. Rodgers, Acta Crystallogr., Sect. A: Found. Crystallogr., 1983, 39, 737–740 CrossRef.
- V. I. Pepekin, Y. N. Matyushin and Y. A. Lebedev, Russ. Chem. Bull., 1974, 23, 1707–1710 CrossRef.
- T. M. Klapötke and D. G. Piercey, Inorg. Chem., 2011, 50, 2732–2734 CrossRef PubMed.
- S. L. Mayo, B. D. Olafson and W. A. Goddard, J. Phys. Chem., 1990, 94, 8897–8909 CrossRef CAS.
- B. M. Rice and J. J. Hare, J. Phys. Chem., 2002, 106, 1770–1783 CrossRef CAS.
- B. M. Rice, Adv. Ser. Phys. Chem., 2005, 16, 335–367 CAS.
- S. L. Mayo, B. D. Olafson and W. A. Goddard, J. Phys. Chem., 1990, 94, 8897–8909 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 









