DOI:
10.1039/C5RA17520H
(Paper)
RSC Adv., 2015,
5, 93591-93606
Synthesis and characterization of novel electrochromic poly(amide-imide)s with N,N′-di(4-methoxyphenyl)-N,N′-diphenyl-p-phenylenediamine units†
Received
29th August 2015
, Accepted 25th October 2015
First published on 28th October 2015
Abstract
We developed a new efficient procedure for the synthesis of a bistriphenylamine diamine monomer, N,N′-bis(4-aminophenyl)-N,N′-bis(4-methoxyphenyl)-1,4-phenylenediamine (3). A new dicarboxylic acid monomer bearing two built-in imide rings, namely N,N′-bis(trimellitimidophenyl)-N,N′-bis(4-methoxyphenyl)-1,4-phenylenediamine (4), was synthesized from the condensation of diamine 3 with two equivalent amounts of trimellitic anhydride. Several novel electroactive poly(amide-imide)s (PAIs) containing N,N′-di(4-methoxyphenyl)-N,N′-diphenyl-p-phenylenediamine [TPPA(OMe)2] units have been prepared by the phosphorylation polyamidation reactions from diamine 3 with four imide ring-preformed dicarboxylic acids or from diimide-diacid 4 with 4,4′-oxydianiline and diamine 3, respectively. All the PAIs were readily soluble in many organic solvents and could be solution-cast into tough and flexible polymer films. These PAIs exhibited glass-transition temperatures (Tgs) in the range 206–292 °C, and most of them did not show significant weight-loss before 450 °C. The PAI films revealed reversible electrochemical oxidation processes accompanied with strong color changes from the pale yellow neutral state to yellowish green and deep blue oxidized. Incorporating the TPPA(OMe)2 unit on the amide side of PAIs led to lower oxidation potentials and higher redox and electrochromic stability.
1. Introduction
Electrochromism is defined as a reversible change in optical absorption (or transmittance) and color change resulting from the redox of the material in response to an externally applied potential by electrochemical means which has stimulated the interest of scientists over the past few decades.1 Traditionally, interest in electrochromic materials has been directed towards optical changes in the visible region, leading to many technological applications such as variable reflectance mirrors, smart windows, and electrochromic displays.2–4 One of the main use of electrochromic materials is in smart windows for car and buildings5 and in automatic-dimming rear-view mirrors.6 Recent high-profile commercialization of electrochromic materials includes the Boeing 787 Dreamliner windows manufactured by Gentex.7 Potential applications in information storage,8 electrochromic displays,9 and adaptive camouflages10 can also be envisioned. There are several chemical systems that are intrinsically electrochromic, such as transition metal oxides (especially tungsten oxide), inorganic coordination complexes, small organic molecules, and conjugated polymers (polyanilines, polypyrroles, and polythiophenes).1–4 Electrochromic applications based on π-conjugated polymers become popular owing to their ease of colour-tuning, fast switching time, and high contrast ratios.11–15 Studies from the Reynolds research group have shown a wide variety of conjugated polymers with colour changes that cover the entire visible spectrum.16–20
Triarylamine derivatives are well known for photo- and electroactive properties that find optoelectronic applications as photoconductors, hole-transporters, and light-emitters.21–23 During the last decade, Liou's and our research groups have developed a number of high-performance polymers, such as aromatic polyamides and polyimides, carrying the triarylamine unit as an electrochromic functional moiety.24–32 Our strategy was to synthesize the triarylamine-containing monomers such as diamines and dicarboxylic acids that were then reacted with the corresponding co-monomers through conventional polycondensation techniques. The obtained polymers possessed characteristically well-defined structures, high molecular weights, and high thermal stability. Because of the incorporation of packing-disruptive, propeller-shaped triarylamine units along the polymer backbone, almost all the polyamides and some of the polyimides exhibited good solubility in polar organic solvents. They could form uniform, transparent amorphous thin films by solution casting and spin-coating methods. This is advantageous for their ready fabrication of large-area, thin-film devices.
Aromatic poly(amide-imide)s (PAIs) possess balanced characteristics between polyamides and polyimides such as high thermal stability and good mechanical properties together with ease of processability.33 Since we successfully applied the Yamazaki–Higashi phosphorylation reaction34 to the direct synthesis of high-molecular-weight PAIs from the imide ring-preformed dicarboxylic acids and aromatic diamines using triphenyl phosphite (TPP) and pyridine as condensing agents, this efficient synthetic route has proved to exhibit significant advantages in preparing operations as compared with conventional acid chloride or isocyanate methods.35 Thus, many novel PAIs have been readily prepared by this convenient technique in our and other laboratories.36 Furthermore, this synthetic procedure can offer us the option of the incorporation of specific functionalities between amide or imide groups in the PAI backbone. The incorporation of such functional groups may provide a method of controlling certain physical properties or special functions of the resulting PAIs.
The anodic oxidation pathways of triphenylamine (TPA) derivatives were well studied.37 The electrogenerated cation radical of TPA is not stable and could dimerize to form tetraphenylbenzidine by tail to tail coupling with loss of two protons per dimer. When the phenyl groups were incorporated by electron-donating substituents such as tert-butyl and methoxy groups at the para-position of TPA, the coupling reactions were greatly prevented by affording stable cationic radicals and lowering the oxidation potentials.38 The redox property, electron-transfer process, and multi-colour electrochromic behaviour of the polymers bearing N,N,N′,N′-tetraphenyl-1,4-phenylenediamine (TPPA) segments are interesting for electro-optical applications.39–44 As reported by Yen and Liou in 2009, a methoxy-blocked TPPA-based diamine monomer N,N′-bis(4-aminophenyl)-N,N′-bis(4-methoxyphenyl)-1,4-phenylenediamine (compound 3 as shown in Scheme 1) was synthesized and used for the preparation of highly organosoluble and stable visible/near-infrared electrochromic aromatic polyamides.45 However, the polyimides based on diamine 3 were less soluble in organic solvents as compared to their polyamide counterparts.46 Unless bridged dianhydrides such as ODPA, DSDA, and 6FDA were used, the polyimides derived from rigid dianhydrides such as PMDA were sparingly soluble in organic solvents. As a continuation of our efforts in developing easily processable high-performance functional polymers with the TPPA segment, the present study describes a new convenient and efficient synthetic route of the TPPA(OMe)2-diamine 3 and the synthesis and characterization of its derived novel TPPA(OMe)2-containing PAIs. The PAIs are expected to exhibit good solubility while preserve high thermal stability and good redox activity. The effects of the location of TPPA(OMe)2 (i.e., on the imide side or the amide side) on the electrochemical and electrochromic properties of the PAIs are also investigated.
 |
| | Scheme 1 Synthetic route to the TPPA(OMe)2-diamine monomer 3. | |
2. Experimental section
2.1. Materials
The diimide-diacid monomers 5a and 5b and monoimide-diacid 5c and 5d were prepared according to the previously reported procedures.35 4,4′-Oxydianiline (ODA) was used as received from Tokyo Chemical Industry (TCI). N-Methyl-2-pyrrolidone (NMP) (Tedia) was dried over calcium hydride for 24 h, distilled under reduced pressure, and stored over 4 Å molecular sieves in a sealed bottle. Triphenyl phosphite (TPP) (TCI) was used as received from the supplier. Commercially obtained calcium chloride (CaCl2) was dried under vacuum at 180 °C for 3 h prior to use. Tetrabutylammonium perchlorate (Bu4NClO4) (TCI) was recrystallized twice from ethyl acetate under nitrogen atmosphere and then dried in vacuo prior to use. All other reagents and solvents were used as received from commercial sources.
2.2. Monomer synthesis
2.2.1. N,N′-Bis(4-methoxyphenyl)-1,4-phenylenediamine (1). Hydroquinone (22.02 g, 0.2 mol), p-anisidine (54.2 g, 0.44 mol), iodine (5.08 g, 0.04 mol), and 50 mL of xylene were placed in a 100 mL round-bottomed flask equipped with a short path distillation head. The mixture was heated to reflux for 20 h under nitrogen with removal of water. After that, the mixture was poured into 120 mL of ethanol, and the precipitated product was filtered and washed thoroughly with ethanol. Recrystallization from toluene afforded 34.3 g (57% yield) of pale purple crystals with a melting point of 196–199 °C (by DSC at a heating rate of 10 °C min−1). IR (KBr): 3386 cm−1 (N–H stretch). 1H NMR (600 MHz, DMSO-d6, δ, ppm): 3.68 (s, 6H, –OCH3), 6.80 (s, J = 9.0 Hz, 4H, Hc), 6.88 (s, 4H, Ha), 6.91 (d, J = 9.0 Hz, 4H, Hb), 7.46 (s, 2H, N–H).
2.2.2. N,N′-Bis(4-methoxyphenyl)-N,N′-bis(4-nitrophenyl)-1,4-phenylenediamine (2). In a 250 mL round-bottom flask equipped with a stirring bar, a mixture of 9.6 g (0.03 mol) of N,N′-bis(4-methoxyphenyl)-1,4-phenylenediamine (1), 9.3 g (0.066 mol) of p-fluoronitrobenzene, and 10.1 g of cesium fluoride (CsF) in 100 mL of dimethyl sulfoxide (DMSO) was heated at 150 °C for about 18 h. After cooling, the mixture was poured into 500 mL of methanol, and the precipitated product was collected by filtration. Recrystallization from DMF/methanol yielded 10.5 g (62%) of red crystals with a melting point of 228–232 °C (by DSC). IR (KBr): 1585, 1311 cm−1 (–NO2 stretch). 1H NMR (600 MHz, CDCl3, δ, ppm): 3.84 (s, 6H, –OCH3), 6.88 (d, J = 9.0 Hz, 4H, Hd), 6.95 (d, J = 9.0 Hz, 4H, Hc), 7.14 (s, 4H, Ha), 7.15 (d, J = 9.0 Hz, 4H, Hb), 8.03 (d, J = 9.0 Hz, 4H, He).
2.2.3. N,N′-Bis(4-aminophenyl)-N,N′-bis(4-methoxyphenyl)-1,4-phenylenediamine (3). In a 500 mL three-neck round-bottomed flask equipped with a stirring bar under nitrogen atmosphere, 2.32 g (4.12 mmol) of dinitro compound 2 and 0.1 g of 10% Pd/C were dispersed in 250 mL of ethanol. The suspension solution was heated to reflux, and 2 mL of hydrazine monohydrate was added slowly to the mixture. After a further 18 h of reflux, the solution was filtered to remove Pd/C, and the filtrate was cooled under nitrogen atmosphere. Yellowish green crystals grew during the cooling process of the filtrate. The precipitated crystals were collected by filtration and dried in vacuo at 80 °C to give 1.70 g (82% in yield) of pure diamino compound 3; mp = 199–201 °C (by DSC). IR (KBr): 3450, 3365 cm−1 (–NH2 stretch). 1H NMR (600 MHz, DMSO-d6, δ, ppm): 3.68 (s, 6H, –OCH3), 4.92 (s, 4H, –NH2), 6.53 (d, J = 8.4 Hz, 4H, He), 6.68 (s, 4H, Ha), 6.76 (d, J = 8.4 Hz, 4H, Hd), 6.80 (d, J = 9.0 Hz, 4H, Hc), 6.87 (d, J = 9.0 Hz, 4H, Hb). 13C NMR (150 MHz, DMSO-d6, δ, ppm): 55.1 (–OCH3), 114.5 (C5), 114.9 (C9), 121.7 (C1), 123.8 (C4), 126.7 (C8), 136.4 (C7), 141.6 (C3), 142.0 (C2), 145.1 (C10), 154.1 (C6).
2.2.4. N,N′-Bis(trimellitimidophenyl)-N,N′-bis(4-methoxyphenyl)-1,4-phenylenediamine (4). A flask was charged with 1.92 g (10 mmol) of trimellitic anhydride, 2.51 g (5 mmol) of diamine 3, and 60 mL of glacial acetic acid. The heterogeneous mixture was stirred for 1 h at room temperature and then refluxed at 120 °C for 12 h. Then, the mixture was poured into 200 mL of methanol, and the precipitate was collected by filtration and washed thoroughly with methanol to remove acetic acid. The product was washed several times with hot water and then dried in vacuum to afford 3.49 g (82% yield) of the diimide-diacid monomer 4 as red-brown powder; mp = 332–336 °C. The product could be directly used for the subsequent polymerization reaction without any further purification. IR (KBr): 2700–3500 cm−1 (carboxyl O–H stretch), 1776, 1725 cm−1 (imide and carboxyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch). 1H NMR (600 MHz, DMSO-d6, δ, ppm): 13.57 (a broad hump, 2H, –COOH), 3.76 (s, 6H, –OCH3), 6.96–6.98 (two overlapped doublets, 8H, Hc+d), 7.05 (s, 4H, Ha), 7.14 (d, J = 9.0 Hz, 4H, Hb), 7.27 (d, J = 9.0 Hz, 4H, He), 8.05 (d, J = 7.8 Hz, 2H, Hf), 8.28 (d, J = 1.2 Hz, 2H, Hh), 8.39 (dd, J = 7.8, 1.2 Hz, 2H, Hg).
O stretch). 1H NMR (600 MHz, DMSO-d6, δ, ppm): 13.57 (a broad hump, 2H, –COOH), 3.76 (s, 6H, –OCH3), 6.96–6.98 (two overlapped doublets, 8H, Hc+d), 7.05 (s, 4H, Ha), 7.14 (d, J = 9.0 Hz, 4H, Hb), 7.27 (d, J = 9.0 Hz, 4H, He), 8.05 (d, J = 7.8 Hz, 2H, Hf), 8.28 (d, J = 1.2 Hz, 2H, Hh), 8.39 (dd, J = 7.8, 1.2 Hz, 2H, Hg).
2.3. Synthesis of PAIs
The synthesis of PAI 7a was used as an example to illustrate the general synthetic route to produce the PAIs. A mixture of 0.5105 g (0.6 mmol) of diimide-diacid monomer 4, 0.1201 g (0.6 mmol) of ODA, 0.1 g of anhydrous calcium chloride, 0.6 mL of triphenyl phosphite (TPP), 0.15 mL of pyridine, and 0.6 mL of NMP was heated with stirring at 120 °C for 3 h. The resulting polymer solution was poured slowly into 150 mL of methanol giving rise to a tough, fibrous precipitate. The precipitated product was collected by filtration, washed thoroughly with hot water and methanol, and then dried to give PAI 7a. The inherent viscosity of the polymer was 0.76 dL g−1, measured in DMAc at a concentration of 0.5 g dL−1 at 30 °C. The IR spectrum of 7a (film) exhibited characteristic amide absorption bands at 3280 cm−1 (amide N–H stretch) and 1668 cm−1 (amide carbonyl stretch), together with the imide absorption bands at 1778 cm−1 (asymmetrical CO stretch) and 1720 cm−1 (symmetrical CO stretch).
2.4. Preparation of the PAI films
A solution of the polymer was made by dissolving about 0.6 g of the PAI sample in 9 mL of hot DMAc. The homogeneous solution was poured into a 7 cm glass Petri dish, which was placed in a 90 °C oven overnight for the slow release of the solvent. The cast film was then released from the glass substrate and was further dried in vacuo at 160 °C for 8 h. The obtained films were about 75 μm in thickness and were used for thermal analyses.
2.5. Instrumentation and measurements
Infrared (IR) spectra were recorded on a Horiba FT-720 FT-IR spectrometer. 1H and 13C NMR spectra were measured on a Bruker Avance III HD-600 MHz NMR spectrometer with tetramethylsilane as an internal standard. The inherent viscosities were determined with a Cannon-Fenske viscometer at 30 °C. Thermogravimetric analysis (TGA) was performed with a Perkin-Elmer Pyris 1 TGA instrument. Experiments were carried out on approximately 4–6 mg of film samples heated in flowing nitrogen or air (flow rate = 40 cm3 min−1) at a heating rate of 20 °C min−1. DSC analyses were performed on a Perkin-Elmer DSC 4000 differential scanning calorimeter at a scan rate of 20 °C min−1 in flowing nitrogen. Electrochemistry was performed with a CHI 750A electrochemical analyzer. Cyclic voltammetry was conducted with the use of a three-electrode cell in which ITO (polymer film area about 0.8 cm × 1.25 cm) was used as a working electrode. A platinum wire was used as an auxiliary electrode. All cell potentials were taken with the use of a home-made Ag/AgCl, KCl (sat.) reference electrode. Ferrocene was used as an external reference for calibration (+0.48 V vs. Ag/AgCl). Voltammograms are presented with the positive potential pointing to the left and with increasing anodic currents pointing downwards. Spectroelectrochemistry analyses were carried out with an electrolytic cell, which was composed of a 1 cm cuvette, ITO as a working electrode, a platinum wire as an auxiliary electrode, and a Ag/AgCl reference electrode. Absorption spectra in the spectroelectrochemical experiments were measured with an Agilent 8453 UV-Visible diode array spectrophotometer.
3. Results and discussion
3.1. Monomer synthesis
The TPPA(OMe)2-diamine monomer 3 was synthesized by a three-step reaction sequence as shown in Scheme 1. In the first step, N,N′-bis(4-methoxyphenyl)-1,4-phenylenediamine (1) was synthesized by condensation of hydroquinone with p-anisidine in the presence of small amounts of iodine according to the Knoevenagel synthesis of aromatic secondary amines.47 In the second step, the intermediate dinitro compound, N,N′-bis(4-methoxyphenyl)-N,N′-bis(4-nitrophenyl)-1,4-phenylenediamine (2) was obtained by the nucleophilic aromatic fluoro-displacement reaction of p-fluoronitrobenzene with the secondary amine 1 in the presence of cesium fluoride (CsF). Finally, the target diamine monomer 3 was prepared by hydrazine Pd/C-catalyzed reduction of dinitro compound 2. The yield of crude product in each step was generally higher than 80%. This strategy for the preparation of diamine 3 seems to be more convenient and more efficient than that using Ullmann reaction reported by Yen and Liou.45
Fig. S1 (ESI†) illustrates FT-IR spectra of compounds 1 to 3. The IR spectrum of compound 1 gives rise to a sharp, medium absorption at about 3386 cm−1 ascribed to the secondary amine N–H stretching vibration. The dinitro compound 2 shows the characteristic absorption of nitro groups at around 1585 and 1311 cm−1 (–NO2 asymmetric and symmetric stretching). After reduction, the characteristic absorptions of the nitro group disappeared and the amino group shows the typical –NH2 stretching absorption pair at 3450 and 3365 cm−1 as shown in the IR spectrum of diamine 3. The 1H NMR spectrum of the secondary amine 1 is illustrated in Fig. S2 (ESI†). The aromatic NH groups appear at 7.46 ppm as a sharp singlet, and the tall singlet at 3.68 ppm is attributed to the methoxy groups. The hydrogens on the benzene rings appear in the range of 6.79–6.93 ppm. The 1H and H–H COSY NMR spectra of the dinitro monomer 2 are compiled in Fig. S3 (ESI†). The 1H, H–H COSY, 13C and C–H HMQC NMR spectra of the diamine monomer 3 are compiled in Fig. 1 and 2, respectively. These 1H and 13C NMR spectra are essentially identical to those reported in literature,45 where the intermediate dinitro compound 2 has been prepared by a different synthetic route. Only a slight difference in chemical shifts of the resonance peaks was observed possibly due to the use of different test solvents. The 1H NMR spectra confirm that the nitro groups have been completely transformed into amino groups by the high field shift of the aromatic protons, especially for protons He ortho to the amino group, and the resonance signal at about 4.92 ppm corresponding to the aryl primary amino protons. The assignments of all hydrogen and carbon atoms are assisted by two-dimensional (2-D) NMR spectra and are consistent with the proposed structures of compounds 2 and 3.
 |
| | Fig. 1 (a) 1H and (b) H–H COSY NMR spectra of diamine 3 in DMSO-d6. | |
 |
| | Fig. 2 (a) 13C and (b) C–H HMQC NMR spectra of diamine 3 in DMSO-d6. | |
The new diimide-diacid monomer 4 containing the TPPA(OMe)2 unit was synthesized by condensation of diamine 3 with two molar equivalent amount of trimellitic anhydride (TMA) in refluxing glacial acetic acid (Scheme 2). The structure of diimide-diacid 4 was confirmed by IR and 1H NMR spectroscopies. As shown in Fig. S4 (ESI†), the IR spectrum of diimide-diacid 4 shows absorption bands around 2700–3500 cm−1 (–OH stretch, carboxylic acid), 1776 (imide CO asymmetrical stretching), and 1725 cm−1 (imide CO symmetrical stretching and carboxyl CO stretching), confirming the presence of imide ring and carboxylic acid groups in the structure. Fig. 3(a) shows the high-resolution 1H NMR spectrum of 4 in deuterated dimethyl sulfoxide (DMSO-d6). Assignments of each proton are assisted by the H–H COSY NMR spectrum (Fig. 3(b)), and the spectra agree well with the proposed molecular structure of 4.
 |
| | Scheme 2 Synthesis of diimide-diacid monomer 4. | |
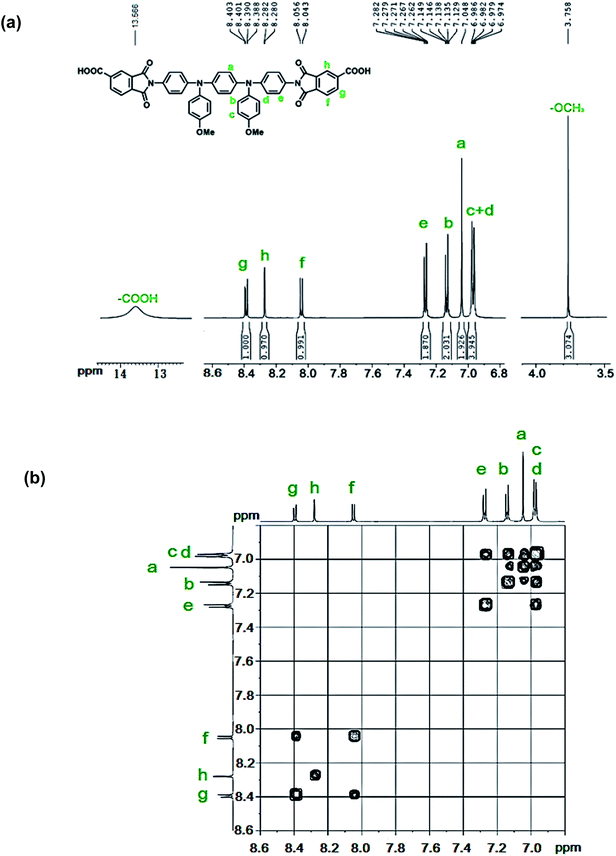 |
| | Fig. 3 (a) 1H and (b) H–H COSY NMR spectra of diimide-diacid monomer 4 in DMSO-d6. | |
3.2. Synthesis of PAIs
According to the phosphorylation polyamidation technique described by Yamazaki and co-workers,34 a series of novel PAIs 6a–6d were synthesized from various combinations of diamine 3 with imide ring-preformed dicarboxylic acids shown in Scheme 3 by using triphenyl phosphite (TPP) and pyridine (Py) as condensing agents. Another type of PAIs 7a and 7b were prepared from ODA and diamine 3, respectively, with the newly synthesized diimide-diacid 4 by a similar synthetic technique (Scheme 4). All the polymerization reactions proceeded homogeneously throughout the reaction and afforded clear, highly viscous polymer solutions. All the polymers precipitated in a tough, fiber-like form when the resulting polymer solutions were slowly poured under stirring into methanol. The obtained PAIs had inherent viscosities in the range of 0.72–0.84 dL g−1, as listed in Table 1. All the polymers can be solution-cast into flexible and transparent films, indicating the formation of high molecular weight polymers.
 |
| | Scheme 3 Synthesis of PAIs 6a–6d. | |
 |
| | Scheme 4 Synthesis of PAIs 7a and 7b. | |
Table 1 Inherent viscosity and solubility behavior of PAIs
| Polymer code |
ηinha (dL g−1) |
Solubility in various solventsb,c |
| NMP |
DMAc |
DMF |
DMSO |
m-Cresol |
THF |
| Inherent viscosity measured at a concentration of 0.5 dL g−1 in DMAc at 30 °C. Solvent: NMP: N-methyl-2-pyrrolidone; DMAc: N,N-dimethylacetamide; DMF: N,N-dimethylformamide; DMSO: dimethyl sulfoxide; THF: tetrahydrofuran. The qualitative solubility was tested with 10 mg of a sample in 1 mL of stirred solvent. ++, soluble at room temperature; +−, partially soluble on heating; −−, insoluble even on heating. |
| 6a |
0.84 |
++ |
++ |
++ |
++ |
++ |
−− |
| 6b |
0.79 |
++ |
++ |
++ |
+− |
++ |
+− |
| 6c |
0.75 |
++ |
++ |
++ |
+− |
++ |
−− |
| 6d |
0.80 |
++ |
++ |
++ |
++ |
++ |
+− |
| 7a |
0.76 |
++ |
++ |
++ |
++ |
++ |
+− |
| 7b |
0.72 |
++ |
++ |
++ |
+− |
++ |
−− |
The structures of the PAIs were confirmed by IR and NMR spectroscopy. Fig. S5 (ESI†) shows the IR spectra of PAIs 6a–6d, and Fig. S6† illustrates those of PAIs 7a and 7b. All the PAIs showed the characteristic absorption bands of the amide group at around 3288 cm−1 (N–H stretching) and 1668 cm−1 (amide CO), and the characteristic imide absorption bands at around 1774 cm−1 (asymmetric CO) and 1722 cm−1 (symmetric CO). As a representative example, the 1H and H–H COSY NMR spectra of PAI 7a are compiled in Fig. 4. The proton NMR spectra of another two examples for PAIs 6a and 7b are respectively depicted in Fig. S7 and S8 (ESI†). The specific assignment of individual hydrogen atoms could be made with the aid of 2-D NMR spectra. All the spectra agree well with the proposed repeating units in these PAIs.
 |
| | Fig. 4 (a) 1H and (b) H–H COSY NMR spectra of PAI 7a in DMSO-d6. | |
3.3. Properties of PAIs
3.3.1. Solubility. The qualitative solubility properties of PAIs in several organic solvents at 10% (w/v) are summarized in Table 1. All of the PAIs were readily dissolved in polar aprotic solvents such as NMP, DMAc, and DMF at room temperature. They are also easily soluble in m-cresol. However, most of them are sparingly soluble in DMSO and less polar THF. The high solubility of these PAIs can be attributed to the incorporation of bulky, packing-disruptive, and three-dimensional TPPA(OMe)2 units in the polymer structure, which results in a high steric hindrance for close packing and thus reduces their crystallization tendency and interchain interactions. Therefore, the good solubility to most polar organic solvents makes these polymers potential candidates for practical applications by simple solution processing.
3.3.2. Thermal properties. The thermal properties of all the PAIs were investigated by DSC and TGA techniques. The thermal behavior data of PAIs are summarized in Table 2. Typical TGA and DSC curves of the representative PAI 7b are shown in Fig. 5. The glass-transition temperatures (Tg) of these PAIs could be easily determined by the DSC method; they were observed in the range of 206–292 °C (see Fig. S9 in ESI†). Semi-aromatic PAIs 6b and 6d exhibited relatively lower Tg values (218 and 206 °C, respectively) when compared with wholly aromatic PAIs 6a (Tg = 287 °C) and 6c (Tg = 278 °C) due to the presence of flexible polymethylene segments. PAIs 7a (Tg = 285 °C) and 7b (Tg = 292 °C) also reveal high Tgs because of high aromatic content. All of the aromatic PAIs exhibited a similar TGA pattern with no significant weight loss below 450 °C in air or nitrogen atmosphere. The decomposition temperatures (Td) at which a 10% weight-loss of the PAIs in nitrogen and air were recorded in the range of 474–548 °C and 457–540 °C, respectively. As expected, PAIs 6b and 6d decompose at slightly lower temperatures than other PAIs because of the less stable aliphatic segments. The amount of carbonized residue (char yield) of all the PAIs in nitrogen atmosphere was more than 58% at 800 °C. The high char yields of the aromatic PAIs can be ascribed to their high aromatic content. The thermal analysis results revealed that these PAIs generally exhibited good thermal stability, which in turn is beneficial to increase the service time in device application and enhance the morphological stability to the spin-coated films.
Table 2 Thermal properties of PAIs
| Polymer codea |
Tgb (°C) |
Td at 5% weight lossc (°C) |
Td at 10% weight lossc (°C) |
Char yieldd (wt%) |
| N2 |
Air |
N2 |
Air |
| All the polymer film samples were heated at 300 °C for 1 h before all the thermal analyses. Midpoint temperature of the baseline shift on the second DSC heating trace (rate = 20 °C min−1) of the sample after quenching from 400 to 50 °C (rate = 20 °C min−1) in nitrogen. Decomposition temperature at which a 5% or 10% weight loss was recorded by TGA at a heating rate of 20 °C min−1 and a gas flow rate of 20 cm3 min−1. Residual weight% at 800 °C at a scan rate 20 °C min−1 in nitrogen. |
| 6a |
287 |
496 |
488 |
548 |
540 |
68 |
| 6b |
218 |
482 |
465 |
502 |
497 |
63 |
| 6c |
278 |
480 |
465 |
515 |
509 |
67 |
| 6d |
206 |
443 |
414 |
474 |
457 |
58 |
| 7a |
285 |
487 |
491 |
530 |
535 |
69 |
| 7b |
292 |
496 |
483 |
548 |
539 |
71 |
 |
| | Fig. 5 TGA and DSC curves of PAI 7b with a heating rate 20 °C min−1. | |
3.3.3. Electrochemical properties. The electrochemical behavior of the PAIs was investigated by cyclic voltammetry (CV) conducted for the cast films on an ITO-coated glass substrate as working electrode in dry acetonitrile (CH3CN) (for anodic oxidation) containing 0.1 M of Bu4NClO4 as the supporting electrolyte using saturated Ag/AgCl as the reference electrode. The derived oxidation potentials are summarized in Table 3. All the 6 series PAIs exhibit two reversible oxidation redox couples with half-wave potentials (E1/2) in the ranges of 0.60–0.65 V and 0.95–1.00 V, respectively, corresponding to successive one electron removal from the TPPA(OMe)2 units. The CV diagrams of PAI 6d, representative for the other 6 series PAIs, are shown in Fig. 6. PAI 6d exhibits very high electrochemical stability because of the active sites of the TPPA are blocked with methoxy groups. It displays two reversible oxidation processes with oxidation peaks at about 0.60 and 0.95 V, respectively. During the electrochemical oxidation of the PAI films, it was also found that the color of the film changed from pale yellow to yellowish green and then to dark blue. Because of the electrochemistry stability and good adhesion between the polymer thin film and the ITO substrate, 6d still exhibited very high electroactivity after 100 repetitive cyclic scans between 0.0 V to 1.2 V.
Table 3 Optical and electrochemical properties of PAIs
| Polymer |
UV-vis absorptiona (nm) |
Oxidation potentialb (V) |
Optical bandgap Egc (eV) |
HOMOd (eV) |
LUMOd (eV) |
| λmax |
λonset |
Eonset |
EOx11/2 |
EOx21/2 |
| UV-vis absorption maximum and onset for the polymer films. Calculated from first CV scans, versus Ag/AgCl in acetonitrile at a scan rate of 50 mV s−1. Optical bandgap calculated from absorption edge of the polymer film: Eg = 1240/λonset. The HOMO energy levels were calculated from EOx11/2 values from the CV curves and were referenced to ferrocene (4.8 eV relative to the vacuum energy level, EOx1/2 = 0.44 V vs. Ag/AgCl). EHOMO = EOx11/2 + 4.8 − 0.44 (eV); ELUMO = EHOMO − Eg. |
| 6a |
311 |
434 |
0.41 |
0.56 |
0.92 |
2.86 |
4.92 |
2.06 |
| 6b |
310 |
426 |
0.38 |
0.51 |
0.87 |
2.91 |
4.87 |
1.96 |
| 6c |
307 |
421 |
0.34 |
0.48 |
0.84 |
2.94 |
4.84 |
1.90 |
| 6d |
320 |
413 |
0.35 |
0.50 |
0.86 |
3.00 |
4.86 |
1.86 |
| 7a |
345 |
433 |
0.49 |
0.63 |
1.02 |
2.86 |
4.99 |
2.13 |
| 7b |
315 |
422 |
0.35 |
0.49 |
0.88 |
2.94 |
4.85 |
1.91 |
 |
| | Fig. 6 Cyclic voltammograms of the cast film of PAI 6d on the ITO-coated glass substrate in 0.1 M Bu4NClO4/CH3CN at a scan rate of 50 mV s−1. | |
The first anodic CV scans of PAIs 6a and 7a with an isomeric repeat unit are compared in Fig. 7. PAI 6a revealed a lower oxidation potential (Eonset = 0.41 V, Epa = 0.65 V) as compared to its isomeric PAI 7a (Eonset = 0.49 V, Epa = 0.74 V) because its TPPA(OMe)2 unit is located in the amide side, which should be more easily oxidized than that in the imide side. PAI 7b contains the TPPA(OMe)2 segment in both amide and imide sides and is expected to show more oxidation processes. However, as shown in Fig. 8, PAI 7b only showed two redox couples similar to other PAIs. The peaks are broader than those of other PAIs due to close overlapping of the redox waves.
 |
| | Fig. 7 Cyclic voltammograms of the cast films of PAIs 6a and 7a on the ITO-coated glass substrate in 0.1 M Bu4NClO4/CH3CN at a scan rate of 50 mV s−1. | |
 |
| | Fig. 8 Cyclic voltammogram of the cast film of 7b on the ITO-coated glass substrate in 0.1 M Bu4NClO4/CH3CN at a scan rate of 50 mV s−1. | |
The redox potentials of the PAIs as well as their respective highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) (versus vacuum) energy levels are summarized in Table 3. The external ferrocene/ferrocenium (Fc/Fc+) redox standard has E1/2 value of 0.44 V vs. Ag/AgCl in acetonitrile. Under the assumption that the HOMO level or called ionization potentials (versus vacuum) for the ferrocene standard was 4.80 eV with respect to the zero vacuum level, the HOMO levels for the PAIs are estimated from the EOx11/2 values of their CV diagrams as 4.84–4.99 eV. The energy gaps estimated from the absorption spectra were then used to obtain the LUMO energy levels of 1.86–2.06 eV. The low ionization potentials suggest an easier hole injection into films from ITO electrodes in electronic device applications.
3.3.4. Spectroelectrochemical and electrochromic properties. Spectroelectrochemical measurements were performed on films of polymers drop-coated onto ITO-coated glass slides immerged in an electrolyte solution. The electrode preparations and solution conditions were identical to those used in the CV experiments. During the test, a three-electrode configuration was used for applying potential to the polymer films in a 0.1 M Bu4NClO4/CH3CN electrolyte solution. When the films were electrochemically oxidized, a strong color change was observed.Fig. 9(a) shows the spectral changes of PAI 6a film upon oxidative scans from 0 to 1.00 V. In the neutral form, PAI 6a exhibited strong absorption at λmax of 311 nm due to the π–π* transitions. As the applied voltage was stepped from 0.0 to 0.65 V, the absorbance at 311 nm decreased, and new peak at 426 nm and a broadband from 985 nm extended into the near-infrared (NIR) region grew up. In the same time, the film turned into yellowish green. We attribute these spectral changes to the formation of a stable monocation radical in the TPPA(OMe)2 moiety. The absorption band in the NIR region is assigned to an intervalence charge-transfer (IV-CT) between states in which the positive charge is centered at different amino centers.48,49 As the applied voltage was raised to 1.00 V, the absorbance at 426 nm decreased and a new broad absorption band at 815 nm started to intensify. Meanwhile, the film changed color from yellowish green to dark blue as shown in the inset of Fig. 9(a). The spectral change of semi-aromatic PAI 6d upon oxidation is very similar to that of 6a (see Fig. S10 in ESI†). For comparison, the spectral change of the cast film of the isomeric PAI 7a on ITO-glass upon oxidative scans is illustrated in Fig. 9(b). In the neutral form, the 7a exhibited strong absorption at λmax of 345 nm. As the applied voltage was stepped from 0.0 to 0.74 V, the absorbance at 345 nm decreased, and new peak at 425 nm and a broadband having its maximum around 980 nm in the NIR region grew up because of the IV-CT effect. In the same time, the film turned from nearly colorless into pale yellow due to the first oxidation process. At a higher applied potential of 1.15 V, the absorbance at 425 nm decreased and a new broad absorption band at 796 nm, a slight blue shift compared with that of isomeric 6a (815 nm), started to intensify, and the film changed color from pale yellow to deep blue (see the inset shown in Fig. 9(b)).
 |
| | Fig. 9 Spectroelectrograms and color changes of (a) PAI 6a (330 ± 50 nm in thickness) and (b) PAI 7a (250 ± 20 nm in thickness) thin films on the ITO-coated glass substrate in 0.1 M Bu4NClO4/CH3CN at various applied voltages (inset: polymer films with about 1.6 μm in thickness). | |
A similar spectral change was observed for PAI 7b at early stage of oxidation (Fig. 10). The absorption peak at 423 nm and the IV-CT band at 988 nm grew up when the applied potential was increased to 0.76 V. This result illustrates that the first two oxidations of the TPPA(OMe)2 unit in both the amide and imide sides occurred almost simultaneously. Further increase of the applied potential over 0.95 V led to the increase of the absorption intensity at about 585 and 801 nm. This absorption change may be explained by the occurrence of the third oxidation. Upon further increase of the potential to 1.12 V, the intensity of the absorption band at 423 nm decreased and the bands around 585 and 801 nm further intensified. It was also found the IV-CT band in the NIR region diminished gradually, implying the occurrence of the fourth oxidation. When the polymer film of 7b was electrochemically oxidized, its color changed from pale yellow to yellowish green, cyan, and deep blue, as can be seen from the inset of Fig. 10.
 |
| | Fig. 10 Spectroelectrograms and color changes of PAI 7b thin film (320 ± 30 nm in thickness) on the ITO-coated glass substrate in 0.1 M Bu4NClO4/CH3CN (inset: polymer film with 1.7 μm in thickness). | |
3.3.5. Electrochromic switching and stability. The stability, response time, and color efficiency are the key parameters for an electroactive polymer film to be amenable for usage in optical and electrochromic devices, thus the electrochromic switching studies were further measured. Electrochromic switching studies for the PAIs were performed to monitor the percent transmittance changes (ΔT%) as a function of time at their absorption maximum (λmax) and to determine the response time by stepping potential repeatedly between the neutral and oxidized states. The polymer film was cast onto an ITO-coated glass slides in the same manner as described earlier, and the active area of the polymer film on ITO glass is about 1 cm2. When the films were switched, the absorbance at the given wavelength was monitored as a function of time with UV-vis-NIR spectroscopy. Fig. 11 and 12 depict the optical transmittance of 6a film as a function of time at 426 and 815 nm by applying square-wave potential steps between 0 and 0.65 V and between 0 and 1.00 V for a switching time of 15 s. Generally, the charge passed at 90% of the full optical switch was used to evaluate the response time since the naked eye could not distinguish the color change after this point. As shown in Fig. 11(a), the polymer film of 6a still exhibited very good electrochromic response after over 20 cyclic scans between 0 and 0.65 V. PAI 6a film attained 90% of a complete coloring and bleaching in 4.8 and 2.1 s, respectively. The optical contrast measured as ΔT% between neutral pale yellow and oxidized yellowish green states was found to be 33% at 426 nm [Fig. 11(b)]. When 6a was switched between neutral and fully oxidized states (the applied potential switched between 0 and 1.00 V), almost no optical contrast loss was observed during switching in the first twenty cycles [Fig. 12(a)]. The response times for coloring and bleaching are recorded as 3.6 and 4.1 s, respectively. The optical contrast between neutral pale yellow and oxidized deep blue states was found to be 73% at 815 nm [Fig. 12(b)]. In addition, the electrochromic coloration efficiency (CE; η) can be calculated via optical density using the following equation:1,2 η = ΔOD/Qd, where ΔOD is the optical absorbance change, and Qd (mC cm−2) is the inject/ejected charge during a redox step. On the basis of this equation, the CE values of the 6a film estimated from the data were found to be 318 cm2 C−1 at 426 nm and 375 cm2 C−1 at 815 nm.
 |
| | Fig. 11 Potential step absorptiometry of the cast films of 6a on the ITO-glass slide (coated area ∼ 1 cm2) (in CH3CN with 0.1 M Bu4NClO4 as the supporting electrolyte) by applying a potential step: (a) optical switching at potential 0.00 V ⇔ 0.70 V (20 cycles) and a switching time of 15 s, monitored at λmax = 426 nm; (b) the 1st cycle transmittance change for the 6a thin film. | |
 |
| | Fig. 12 Potential step absorptiometry of the cast films of 6a on the ITO-glass slide (coated area ∼ 1 cm2) (in CH3CN with 0.1 M Bu4NClO4 as the supporting electrolyte) by applying a potential step: (a) optical switching at potential 0.00 V ⇔ 1.00 V (20 cycles) and a switching time of 15 s, monitored at λmax = 815 nm; (b) the 1st cycle transmittance change for the 6a thin film. | |
The potential step absorptiometry of the 6d film is included in the ESI.† Fig. S11 and S12† depict the optical transmittance of 6d film as a function of time at 430 and 817 nm by applying square-wave potential steps between 0.00 and 0.70 V for a residence time of 7 s and between 0.00 and 1.05 V for a residence time of 12 s. As shown in Fig. S11(a),† the polymer film of 6d still exhibited very good electrochromic response after over 50 cyclic scans between 0 and 0.70 V. PAI 6d film attained 90% of a complete coloring and bleaching in 2.2 and 1.4 s, respectively. The optical contrast measured as ΔT% between neutral pale yellow and oxidized yellowish green states was found to be 36% at 430 nm [Fig. S11(b)†]. When 6d was switched between neutral and fully oxidized states (the applied potential switched between 0 and 1.05 V), only a very small optical contrast loss was observed during switching [Fig. S12(a)†]. The response times for coloring and bleaching are similar to those switching between neutral and first oxidized state. The optical contrast measured as ΔT% between neutral pale yellow and oxidized deep blue states was found to be 71% at 817 nm [Fig. S12(b)†].
The stability and switching behavior of the 7a film at λmax = 425 nm and 796 nm were investigated by monitoring the electrochromic contrast (ΔT%) of the thin film upon repeated square-wave potential steps between 0 and 0.75 V and between 0 and 1.15 V for a pulse width of 15 s. The results are shown in Fig. 13 and 14, respectively. In this case, a response time required for 90% full-transmittance change of 3.9 s for the yellowish green coloring step and 2.2 s for the bleaching step at 425 nm and 3.5 s for the blue coloring step and 2.4 s for the bleaching step at 796 nm. In addition, the optical contrast measured (ΔT%) recorded at neutral and oxidized forms was found to be 32% at 425 nm and 65% at 796 nm. The CE values of the 7a film were calculated as 284 cm2 C−1 at 425 nm and 319 cm2 C−1 at 796 nm (Table 4) by chronoamperometry. The 7a film showed a larger loss of electrochromic contrast as compared to the 6a film with the isomeric repeat unit in the first 20 scans. The less stability of the 7a film might be attributable to the strong electron-withdrawing effect of the imide group. In contrast, the 6a film exhibited excellent electrochromic stability because the TPPA(OMe)2 unit is located in the amide side. The switching results of PAI 7b are included in the ESI.† As shown in Fig. S13,† PAI 7b exhibits good stability in the first 20 full switches, and almost no optical contrast loss after 20 full switches was observed if the polymer film was switched between 0 and 1.15 V (Fig. S14†). This result also indicated that incorporating the TPPA(OMe)2 unit on the amide side of PAIs led to lower oxidation potentials, higher redox reversibility, and better electrochromic performance.
 |
| | Fig. 13 Potential step absorptiometry of the cast film of 7a on the ITO-glass slide (coated area ∼ 1 cm2) (in CH3CN with 0.1 M Bu4NClO4 as the supporting electrolyte) by applying a potential step: (a) optical switching at potential 0.00 V ⇔ 0.75 V (20 cycles) and a switching time of 15 s, monitored at λmax = 425 nm; (b) the 1st cycle transmittance change for the 7a thin film. | |
 |
| | Fig. 14 Potential step absorptiometry of the cast film of 7a on the ITO-glass slide (coated area ∼ 1 cm2) (in CH3CN with 0.1 M Bu4NClO4 as the supporting electrolyte) by applying a potential step: (a) optical switching at potential 0.00 V ⇔ 1.15 V (20 cycles) and a switching time of 15 s, monitored at λmax = 796 nm; (b) the 1st cycle transmittance change for the 7a thin film. | |
Table 4 Electrochromic properties of PAIs
| Polymer |
λmaxa (nm) |
ΔT% |
Response timeb |
ΔODc |
Qdd (mC cm−2) |
CEe (cm2 C−1) |
| tc (s) |
tb (s) |
| Wavelength of absorption maximum. Time for 90% of the full-transmittance change. Optical density change (ΔOD) = log[Tbleached/Tcolored], where Tcolored and Tbleached are the maximum transmittance in the oxidized and neutral states, respectively. Qd is ejected charge, determined from the in situ experiments. Coloration efficiency (CE) = ΔOD/Qd. |
| 6d |
430 |
36 |
2.24 |
1.37 |
0.232 |
0.64 |
362 |
| 817 |
71 |
2.70 |
1.34 |
0.709 |
1.76 |
403 |
| 6a |
426 |
33 |
4.83 |
2.11 |
0.235 |
0.74 |
318 |
| 815 |
73 |
3.64 |
4.06 |
0.723 |
1.93 |
375 |
| 7a |
425 |
32 |
3.97 |
2.23 |
0.216 |
0.76 |
284 |
| 796 |
65 |
3.51 |
2.37 |
0.680 |
2.13 |
319 |
| 7b |
423 |
31 |
5.29 |
3.41 |
0.214 |
0.71 |
301 |
| 812 |
75 |
2.19 |
1.97 |
0.755 |
1.95 |
387 |
4. Conclusions
A new synthetic procedure for N,N′-bis(4-amionophenyl)-N,N′-bis(4-methoxyphenyl)-1,4-phenylenediamine was developed. Several novel TPPA(OMe)2-based PAIs have been easily prepared from the imide ring-preformed dicarboxylic acids with the synthesized diamine or from N,N′-bis(trimellitimidophenyl)-N,N′-bis(4-methoxyphenyl)-1,4-phenylenediamine with aromatic diamines via the phosphorylation polyamidation reaction. Because of the presence of TPPA(OMe)2 unit in the main chain, all the polymers exhibited high solubility in many polar aprotic solvents and good film-forming ability. These PAIs also showed moderate to high thermal stability with glass-transition temperatures in the range of 206–292 °C and 10 wt% loss temperatures in excess of 450 °C. All the PAIs displayed at least two reversible redox waves on their anodic cyclic voltammograms. Incorporating the TPPA(OMe)2 unit on the amide side of PAIs led to lower oxidation potentials, higher redox reversibility, and better electrochromic performance. This new PAI family could be good candidates as anodically electrochromic or hole-transporting materials due to their proper oxidation potentials, high electrochemical and electrochromic stability, and easy processability.
Acknowledgements
The financial support from the Ministry of Science and Technology, Taiwan, is greatly appreciated.
Notes and references
- P. M. S. Monk, R. J. Mortimer and D. R. Rosseinsky, Electrochromism: fundamentals and applications, VCH, Weinheim, Germany, 1995 Search PubMed.
- P. M. S. Monk, R. J. Mortimer and D. R. Rosseinsky, Electrochromism and electrochromic devices, Cambridge University Press, Cambridge, UK, 2007 Search PubMed.
- R. J. Mortimer, Chem. Soc. Rev., 1997, 26, 147–156 RSC.
- D. R. Rosseinsky and R. J. Montimer, Adv. Mater., 2001, 13, 783–793 CrossRef CAS.
-
(a) R. D. Rauch, Electrochim. Acta, 1999, 44, 3165–3176 CrossRef;
(b) R. Baetens, B. P. Jelle and A. Gustavsen, Sol. Energy Mater. Sol. Cells, 2010, 94, 87–105 CrossRef CAS.
- http://www.gentex.com and associated pages.
- A. P. Weidner, US Pat., No. 7450294, 2008.
- G. H. Sonmez and B. Sonmez, J. Mater. Chem., 2006, 16, 2473–2477 RSC.
- R. J. Mortimer, A. L. Dyer and J. R. Reynolds, Displays, 2006, 27, 2–18 CrossRef CAS.
- S. Beaupre, A.-C. Breton, J. Dumas and M. Leclerc, Chem. Mater., 2009, 21, 1504–1513 CrossRef CAS.
- G. Sonmez, Chem. Commun., 2005, 5251–5259 RSC.
- A. Patra and M. Bendikov, J. Mater. Chem., 2010, 20, 422–433 RSC.
- P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268–320 CrossRef CAS.
- A. Balan, D. Baran and L. Toppare, Polym. Chem., 2011, 2, 1029–1043 RSC.
- G. Gunbas and L. Toppare, Chem. Commun., 2012, 48, 1083–1101 RSC.
- P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Acc. Chem. Res., 2010, 43, 1395–1407 CrossRef.
- C. M. Amb, P. M. Beaujuge and J. R. Reynolds, Adv. Mater., 2010, 22, 724–728 CrossRef CAS.
- A. L. Dyer, M. R. Craig, J. E. Babiarz, K. Kiyak and J. R. Reynolds, Macromolecules, 2010, 43, 4460–5446 CrossRef CAS.
- C. M. Amb, A. L. Dyer and J. R. Reynolds, Chem. Mater., 2011, 23, 397–415 CrossRef CAS.
- A. L. Dyer, E. J. Thompson and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2011, 3, 1787–1795 CAS.
- M. Thelakkat, Macromol. Mater. Eng., 2002, 287, 442–461 CrossRef CAS.
- Y. Shirota, J. Mater. Chem., 2005, 15, 75–93 RSC.
- Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953–1010 CrossRef CAS.
- S.-H. Cheng, S.-H. Hsiao, T.-H. Su and G.-S. Liou, Macromolecules, 2005, 38, 307–316 CrossRef CAS.
- C.-W. Chang, G.-S. Liou and S.-H. Hsiao, J. Mater. Chem., 2007, 17, 1007–1015 RSC.
- S.-H. Hsiao, G.-S. Liou, Y.-C. Kung and H.-J. Yen, Macromolecules, 2008, 41, 2800–2808 CrossRef CAS.
- Y.-C. Kung and S.-H. Hsiao, J. Mater. Chem., 2010, 20, 5481–5492 RSC.
- H.-M. Wang and S.-H. Hsiao, Polym. Chem., 2010, 1, 1013–1023 RSC.
- Y.-C. Kung and S.-H. Hsiao, J. Mater. Chem., 2011, 21, 1746–1754 RSC.
- H.-J. Yen and G.-S. Liou, Polym. Chem., 2012, 3, 255–264 RSC.
- H.-M. Wang and S.-H. Hsiao, J. Mater. Chem. C, 2014, 2, 1553–1564 RSC.
- J.-H. Wu and G.-S. Liou, Adv. Funct. Mater., 2014, 24, 6422–6429 CrossRef CAS.
- Y. Imai, in Synthesis of Polyamideimides in Polyimide: Fundamentals and Applications, ed. M. K. Ghosh and K. L. Mittal, Marcel Dekker, New York, 1996, pp. 49–70 Search PubMed.
- N. Yamazaki, M. Matsumoto and F. Higashi, J. Polym. Sci., Polym. Chem. Ed., 1975, 13, 1373–1380 CrossRef CAS.
-
(a) C.-P. Yang and S.-H. Hsiao, Makromol. Chem., 1989, 190, 2119–2131 CrossRef CAS;
(b) S.-H. Hsiao and C.-P. Yang, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 1149–1159 CrossRef CAS;
(c) S.-H. Hsiao and C.-P. Yang, Makromol. Chem., 1990, 191, 155–167 CrossRef CAS.
-
(a) D.-J. Liaw, P.-N. Hsu, W.-H. Chen and S.-L. Lin, Macromolecules, 2002, 35, 4669–4676 CrossRef CAS;
(b) H. Behniafar and A. Banihashemi, Polym. Int., 2004, 53, 2020–2025 CrossRef CAS;
(c) H.-M. Wang and S.-H. Hsiao, Polym. Chem., 2010, 1, 1013–1023 RSC;
(d) I. Bacosca, E. Hamciuc, M. Bruma and M. Ignat, React. Funct. Polym., 2011, 71, 905–915 CrossRef CAS;
(e) S.-H. Hsiao, W. Guo, T.-H. Tsai and Y.-T. Chiu, J. Polym. Res., 2014, 21, 391 CrossRef (10 pages).
- E. T. Seo, R. F. Nelson, J. M. Fritsch, L. S. Marcoux, D. W. Leedy and R. N. Adams, J. Am. Chem. Soc., 1966, 88, 3498–3503 CrossRef CAS.
- S.-H. Hsiao, Y.-M. Chang, H.-W. Chen and G.-S. Liou, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4579–4592 CrossRef CAS.
- G.-S. Liou and C.-W. Chang, Macromolecules, 2008, 41, 1667–1674 CrossRef CAS.
- C.-W. Chang, C.-H. Chung and G.-S. Liou, Macromolecules, 2008, 41, 8441–8451 CrossRef CAS.
- C.-W. Chang and G.-S. Liou, J. Mater. Chem., 2008, 18, 5638–5646 RSC.
- H.-M. Wang and S.-H. Hsiao, Polymer, 2009, 50, 1692–1699 CrossRef CAS.
- S.-H. Hsiao, G.-S. Liou and H.-M. Wang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2330–2343 CrossRef CAS.
- H.-M. Wang and S.-H. Hsiao, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 337–351 CrossRef CAS.
- H.-J. Yen and G.-S. Liou, Chem. Mater., 2009, 21, 4062–4070 CrossRef CAS.
- C.-J. Chen, H.-J. Yen, W.-C. Chen and G.-S. Liou, J. Mater. Chem., 2012, 22, 14085–14093 RSC.
- N. P. Buu-Hoi, J. Chem. Soc., 1592, 4346 Search PubMed.
- M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1968, 10, 247–422 CrossRef.
- C. Lambert and G. Noll, J. Am. Chem. Soc., 1999, 121, 8434–8442 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra17520h |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 





















