DOI:
10.1039/C5RA18923C
(Paper)
RSC Adv., 2015,
5, 91846-91854
Evaluation of cobalt oxide, copper oxide and their solid solutions as heterogeneous catalysts for Fenton-degradation of dye pollutants†
Received
15th September 2015
, Accepted 13th October 2015
First published on 15th October 2015
Abstract
A series of CoO, Co0.75Cu0.25O, Co0.5Cu0.5O and CuO nanoparticles were synthesized via the calcination of corresponding oxalates and further examined as catalysts for the heterogeneous Fenton reaction. The structures of the as-prepared oxides were characterized by field emission electron microscopy, transmission electron microscopy, energy dispersive X-ray spectroscopy and X-ray diffraction. The catalytic activity of the oxides was evaluated by the degradation of Congo red. It was found that, among the four catalysts, Co0.5Cu0.5O showed the best catalytic performance. Subsequently, the effects of operating parameters including the substrate concentration, pH, H2O2 concentration and reaction temperature in the catalytic performance of the Co0.5Cu0.5O were systematically studied. Under optimized conditions of catalyst loading = 200 mg L−1, pollutant concentration = 100 mg L−1, H2O2 concentration = 3 wt%, temperature = 30 °C and pH = 9, the Co0.5Cu0.5O catalyst could completely degrade the Congo red within 60 min. The degradation products were analyzed by a liquid chromatography-mass spectrometer and the degradation pathway was revealed. To investigate the catalytic mechanism, the pH and concentrations of H2O2 and metal ions were monitored during the Fenton process. Mechanistic studies revealed that hydroxyl radicals and superoxide radicals derived from the activation of H2O2 molecules by metal centers were mainly responsible for the degradation of Congo red, and that copper ions played a critical role in the superior catalytic performance of the Co0.5Cu0.5O catalyst. The Co0.5Cu0.5O catalyst showed negligible metal leaching and outstanding recyclability, which are highly favorable for the practical application in the Fenton process.
Introduction
Owing to the growth of global population and escalation of environment pollution, recent years have witnessed an ever-increasing demand for supplies of clean and safe water sources. Thus, it is of great importance to explore technologies for water treatment. Nowadays, synthetic dyes constitute a large group of water pollutants due to their massive consumption in the textile, paper, tannery and paint industries.1 The release of synthetic dyes into water sources poses a serious hazard to aquatic ecosystems and human beings.2 So far, a series of physical, chemical, biological, and electrical methodologies have been explored for dye removal.3 Of these methods, advanced oxidation processes (AOPs) are considered to be the most feasible choice. Being one of the most extensively studied AOPs, the Fenton reactions show great application potentials in pollutant degradation because of the high efficiency and simplicity.4 In general, the classic Fenton reaction involves a homogeneous solution of iron (II) ions and hydrogen peroxide. The resulting highly oxidative oxygen-containing species are responsible for pollutant degradation.5 In spite of high efficiency, the homogeneous Fenton reaction also suffers from several drawbacks. The efficiency of the homogeneous Fenton reaction relies on the strong acidic condition (pH < 3) and decreases significantly in the pH range of natural aqueous solutions (pH 5–9).6 In addition, the treatment of large amounts of iron sludge and neutralization of solutions before disposal consume additional time and manpower, leading to increasing overall cost of AOPs.7 To address these issues, efforts have been directed in developing heterogeneous catalysts for the Fenton reaction. Heterogeneous catalysts offer the advantage of facile separation from the effluents. The heterogeneous Fenton reaction mainly takes place at the solid–liquid interface and is able to degrade pollutants in a wider pH range with less metal loss in comparison with the homogeneous Fenton reaction.8
So far, numerous materials have been studied as catalysts for the heterogeneous Fenton reaction.9–16 A series of excellent reviews summarized the heterogeneous Fenton catalysts in the literature.6,17–20 Among the reported heterogeneous catalysts, iron-based catalysts such as goethite,21 hematite,22 magnetite23 and metallic iron24 nanoparticles have attracted considerable attention owing to the properties of redox pairs of FeIII/FeII. It cannot be denied that the FeIII/FeII redox pairs show high efficiency in the activation of H2O2. Nevertheless, the iron-based catalysts generally work in an acidic condition. The stability of iron-based catalysts is still a great concern and iron leaching is always inevitable particularly in the strong acidic solutions.25 In principle, other metal redox pairs such as CuI/CuII,26,27 CoII/CoIII (ref. 28–30) and MnII/MnIII (ref. 31 and 32) are also capable of activating H2O2 to generate oxidative oxygen-containing species. Herein, we synthesized a series of cobalt oxide, copper oxide and their solid solutions, and further investigated the catalytic activity for AOPs. The catalytic activity of as-prepared oxides was evaluated by the degradation of Congo red (CR), a widely used azo dye in textile industries. Although the syntheses of cobalt oxides and copper oxides were widely reported, however, as far as we know, very few studies on application of these oxides as heterogeneous Fenton catalysts are available in the literature.28 The effects of operating parameters including the pH, CR and H2O2 concentrations, and reaction temperature in the degradation of CR were systematically studied. The degradation products were identified and the catalytic mechanism was also explored.
Experimental sections
Synthesis of metal oxides
Metal oxides were obtained from the decomposition of corresponding oxalates as reported by Lua and Wang with slight modifications.33 In brief, oxalates were prepared via precipitation reactions, where nitrates, oxalic acid and ethanol were used as metal precursor(s), precipitant and solvent, respectively. The precipitation reaction was conducted at room temperature and the resulting sediment was aged at 120 °C for 12 h using an autoclave to obtain oxalates. Purified oxalates were calcined at 500 °C for 4 h in air to obtain metal oxides. Four metal oxides including CoO, Co0.75Cu0.25O, Co0.5Cu0.5O and CuO were synthesized.
Characterization methods
Field emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM) were employed to observe the morphology of the samples. The elemental composition of the samples was determined by an energy dispersive X-ray analyzer (EDX) equipped in FESEM. Powder X-ray diffraction (XRD) patterns were recorded by an X-ray diffractometer using Cu Kα (λ = 1.5406 A) radiation at 40 kV and 30 mA. Before the experiments, the X-ray diffractometer was carefully calibrated with a reference of a silicon crystal. All the XRD tests were performed on a continuous scan mode with scan rate of 1° min−1. To determine the peak position and full width at half maximum, the XRD patterns were analyzed using the HighScore software. The d-spacing values of the samples were calculated from the Bragg' law.
Catalytic activity measurements
To evaluate the catalytic activity of as-prepared oxides, a widely used azo dye, CR was selected as a model pollutant. The experimental procedures of CR degradation are as follows: stock solutions of CR with varying concentration were prepared by dissolving analytic-grade CR powder into double-distilled water. The initial pH of CR solution was adjusted by adding 1 M H2SO4 or NaOH solution. A predetermined amount of catalyst sample was added into 100 mL of CR solution under magnetic stirring. The degradation of CR was initiated by adding a given amount of H2O2 solution. The suspension was stirred throughout the experiment. The temperature of the suspension was controlled by a programmed heater. At given time intervals, 2 mL of aliquot was sampled and filtered through a syringe filter to remove catalyst particles. The concentration of CR in the filtrate was analyzed by UV-visible spectroscopy (λmax = 500 nm). The concentrations of dissolved metals in the solutions were analyzed by an atomic absorption spectrometer. The degradation products were determined by a liquid chromatography-mass spectrometer (LC-MS) equipped with a Novpak-C18 column and UV detector. The mobile phase consists of a mixture of methanol and water (75/25, v/v) with a flow rate of 0.8 mL min−1. The concentration of H2O2 was determined using a UV-visible spectroscopy. The details of the procedures are as follows:34 after removing catalyst particles, 10 mL of 3 M H2SO4 and 30 mM Ti2SO4 were added into 2 mL of pollutant solution. The solution was diluted with 40 mL of distilled water. After ca. 10 minutes of shaking, the solution was transferred into a cuvette for analysis (λmax = 406 nm).
Results and discussion
Structural properties
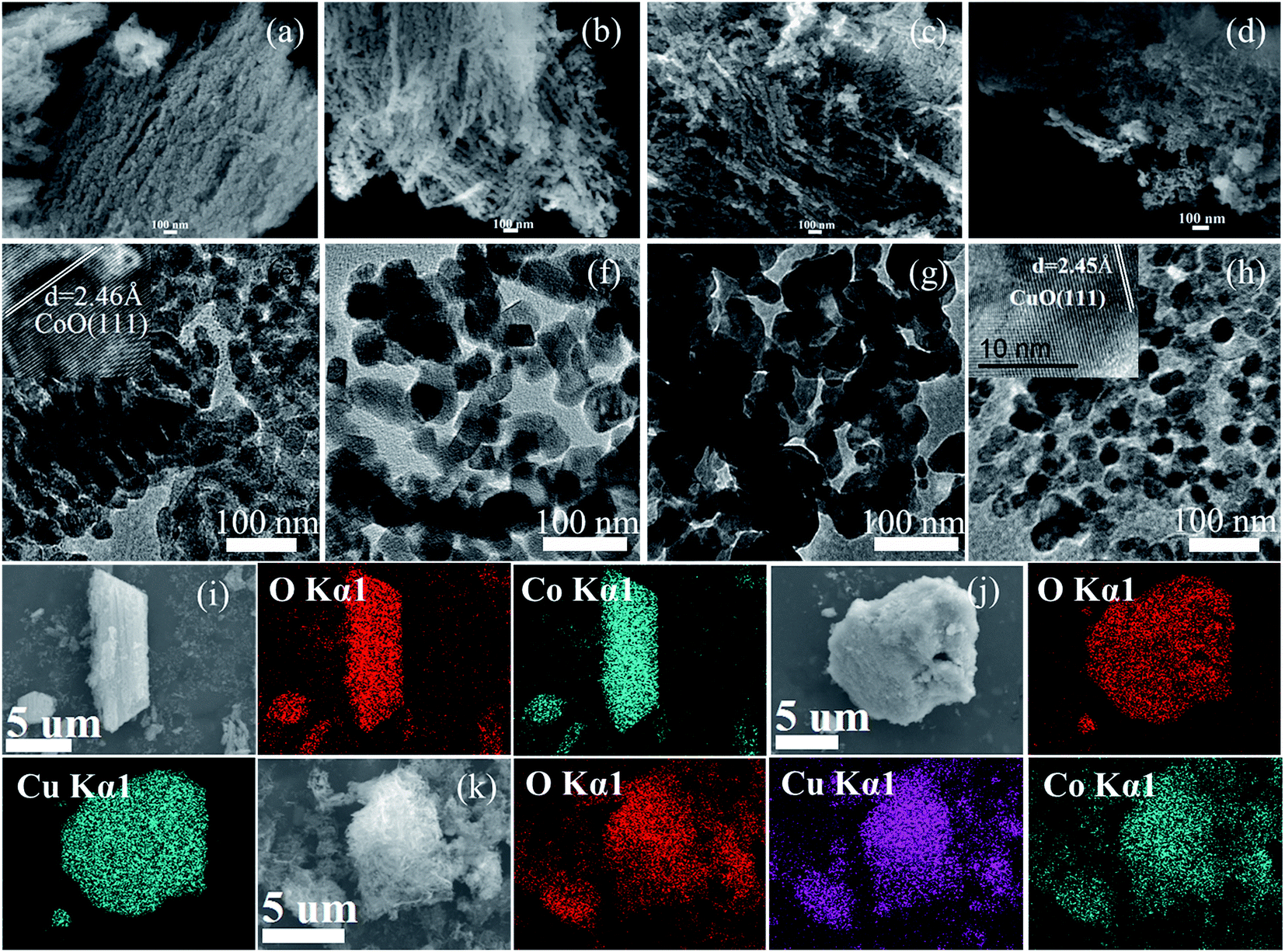
The morphology of as-synthesized metal oxides was observed by FESEM and TEM as shown in Fig. 1. FESEM images (a–d) show that the resulting metal oxides are composed of a number of fine nanoparticles. TEM images (e–h) indicate that these fine nanoparticles exhibit a quasi-spherical structure. Based on the TEM images, for each sample, the sizes of 100 nanoparticles were measured and the average particle sizes of CoO, Co0.75Cu0.25O, Co0.5Cu0.5O and CuO were calculated to be 18, 24, 23 and 16 nm, respectively. The insets shown in Fig. 1e and h present the high-resolution TEM images of CoO and CuO, respectively. The distances of the lattice fringes were carefully measured to be 2.46 and 2.45 nm, corresponding to the spacing values of CoO (111) and CuO (111), respectively. Fig. 1i–k displays the elemental mapping images of the oxides, which clearly resolve the distribution of oxygen (red color), copper (blue color) and cobalt (gray color). Based on the EDX analyses, the actual atomic ratios of CoO, Co0.75Cu0.25O, Co0.5Cu0.5O and CuO are determined to be 0.483![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.517, 0.332:0.127:0.541, 0.246:0.252:0.502, and 0.488:0.512, respectively, which are very close to the nominal composition of the oxides.
:0.517, 0.332:0.127:0.541, 0.246:0.252:0.502, and 0.488:0.512, respectively, which are very close to the nominal composition of the oxides.
 |
| | Fig. 1 FESEM (a–d), TEM (e–h) and elemental mapping images (i–k) of CoO, Co0.75Cu0.25O, Co0.5Cu0.5O and CuO. Insets in (e) and (h) are high-resolution TEM images of CoO and CuO, respectively. | |
To study the crystallographic properties of the oxides, XRD tests were performed and the resulting XRD patterns were carefully analyzed using the HighScore software. Fig. 2a displays the XRD patterns of the oxides. Five diffraction peaks located at 36.5, 42.4, 61.5, 73.7 and 77.6° are clearly noted in the XRD pattern of CoO, which can be well indexed to the diffraction of (111), (200), (220), (311) and (222) planes (Reference code: 01-089-7099), respectively. In the XRD pattern of CuO, these five peaks shift to 36.6, 42.6, 61.8, 74 and 77.9° (reference code: 01-078-0428), respectively. A single set of peaks are presented in the XRD patterns of Co0.75Cu0.25O, and Co0.5Cu0.5O, indicating formation of solid solutions in these binary metal oxides. A careful examination on the XRD patterns can reveal that the peaks of the oxides slightly shift to high angles of 2θ with increasing copper content. For clarity, Fig. 2b displays the enlarged view of the (200) peaks of the oxides. The d-spacing value was accordingly calculated using the Bragg's law and the results are presented in the inset in Fig. 2b. The d(200) spacing values of CoO and CuO are calculated to be 2.13 and 2.12 Å, respectively. The d-spacing value linearly increases with increasing cobalt content, which is well consistent with the Vegard's law.
 |
| | Fig. 2 (a) XRD patterns of the catalysts and (b) enlarged view of peak (200). Inset in (b) is the relationship of spacing value of d(200) and Co/Cu atomic ratio. | |
Catalytic activities
The catalytic activity of as-prepared oxides was evaluated by using CR as a model pollutant. Shown in Fig. 3a, the normalized CR concentration is recorded as a function of time. To check the reproducibility of the results, for each sample, the experiments were repeated for three times and the standard deviates were presented as error bars as shown in Fig. 3a. It is found that the experimental errors in this study are limited and that the results are reproducible. The binary metal oxides, viz., Co0.75Cu0.25O, and Co0.5Cu0.5O exhibit better activity in comparison with the single metal oxides, viz., CuO and CoO. Further inspection reveals that the Co0.75Cu0.25O possesses better activity in the initial 60 min compared with the Co0.5Cu0.5O. However, with further increasing reaction time, the activity of Co0.5Cu0.5O surpasses that of Co0.75Cu0.25O. Within 300 min, the concentration of CR solution decreases from the initial of 100 mg L−1 to 70, 62, 33 and 22 mg L−1 in virtue of CuO, CoO, Co0.75Cu0.25O and Co0.5Cu0.5O catalyst, respectively. Overall, among the four catalysts, the Co0.5Cu0.5O exhibits the highest activity and the activity follows the sequence of CuO < CoO < Co0.75Cu0.25O < Co0.5Cu0.5O. Consequently, the Co0.5Cu0.5O is preferentially selected for further study. Fig. 3b displays the UV-visible spectra of CR solutions treated by Co0.5Cu0.5O catalyst at varying time intervals. It shows that the intensity of the maximum adsorption peaks located at ca. 500 nm decreases with increasing time, indicating the degradation of CR arisen from the catalyzing effects of Co0.5Cu0.5O.
 |
| | Fig. 3 (a) Comparison of four catalysts and (b) UV-vis spectroscopy of CR solutions treated with Co0.5Cu0.5O and H2O2. Experimental conditions: initial pH = 7; catalyst loading = 100 mg L−1; H2O2 concentration = 3%; initial CR concentration = 100 mg L−1; temperature = 30 ± 1 °C. | |
The efficiency of heterogeneous Fenton process is highly dependent on the operating conditions.35–44 The effects of experimental parameters including substrate concentration, H2O2 concentration, pH and temperature in the degradation performance of the Co0.5Cu0.5O catalyst were studied. To investigate the effects of substrate concentration, CR solutions with different initial concentrations varying from 50 to 800 mg L−1 were treated for 24 h and the final CR concentration in the solution (denoted as Ce) was determined. Shown in Fig. 4a, the normalized concentration Ce/C0 monotonically increases with increasing substrate concentration. This is reasonable considering the fact that the degradation of CR pollutant is attributed to the number of oxidative oxygen-containing species. Given a fixed amount of H2O2, the number of the produced oxidative species is limited, leading to increasing Ce/C0 with increasing initial CR concentrations as shown in Fig. 4a. However, it should be pointed out that, high substrate concentrations are always favorable for suppressing the parasitic scavenging reactions occurred in the Fenton reaction, thereby resulting in high efficiency of H2O2.
 |
| | Fig. 4 Effects of (a) substrate concentration, (b) H2O2 concentration and (c) pH of solution in the degradation of CR. Experimental conditions: (a) catalyst loading = 100 mg L−1; H2O2 concentration = 3 wt%; temperature = 30 ± 1 °C; pH = 7; (b) catalyst loading = 100 mg L−1; CR concentration = 100 mg L−1; temperature = 30 ± 1 °C; initial pH = 7; (c) catalyst loading = 100 mg L−1; CR concentration = 100 mg L−1; H2O2 concentration = 3%; temperature = 30 ± 1 °C. | |
It is well known that H2O2 plays a crucial role in the Fenton-degradation of organic pollutants.45,46 Shown in Fig. 4b, the concentration of H2O2 in the pollutant solution indeed affects the degradation of CR. In the absence of H2O2, the CR concentration slightly decreases due to the adsorption of CR to the surface of Co0.5Cu0.5O nanoparticles. With addition of H2O2, the degradation of CR is dramatically accelerated and the degradation performance of the Co0.5Cu0.5O catalyst depends on the H2O2 concentration. The degradation of CR is promoted with increasing H2O2 concentration from 1.5 to 4 wt% but retarded with further increasing H2O2 concentration to 5 wt%, resulting in an optimal H2O2 concentration of 4 wt%. In the Fenton process, the number of oxidative oxygen-containing species is highly dependent on H2O2 concentration. An adequate H2O2 concentration in the CR solution is favorable for increasing the number of oxidative species and facilitating the degradation of CR pollutant. However, when H2O2 is overdosed (>4 wt% in this study), the number of HO˙ decreases as illustrated by following reactions:47
| | | H2O2 + HO˙ → HO2˙ + H2O | (1) |
The pH of pollutant solution affects the surface properties of catalysts as well as formation of oxidative species.48,49 The initial pH of pollutant solution was carefully adjusted (but not buffered) to four different values of 5, 7, 9 and 11 by adding a proper amount of 1 M H2SO4 or NaOH solution. Fig. 4c illustrates the effects of the initial pH in the degradation of CR. Among the four tested initial pH values, the pH of 9 is most favorable for the CR degradation. The Co0.5Cu0.5O catalyst shows similar catalytic performance at the three other pH values. Such a pH-dependence behavior is related to the activation of H2O2 in the Co0.5Cu0.5 catalyst as discussed later. In general, the homogeneous Fenton reaction is conducted at acidic solutions (pH < 3) to achieve high efficiency. However, such a requisite greatly limits the application of the Fenton process because large volumes of pollutant solutions always have neutral or slightly basic pH values (5–9). In this regard, the heterogeneous Fenton reaction mediated by Co0.5Cu0.5O catalyst shows a remarkable advantage because of the working environment.
The effects of reaction temperature in the CR degradation are illustrated in Fig. 5. It is noted that the degradation performance of the Co0.5Cu0.5O catalyst increases with increasing temperature. At 20 °C, the degradation of CR is slow and the CR concentration is still up to 46 mg L−1 after 300 min, which is much higher than those of 22, 7 and 2 mg L−1 at 30, 40 and 50 °C. To reveal the kinetics of the degradation reaction, the experimental data were fitted with the pseudo-first kinetics equation. The apparent rate constant was obtained by plotting ln(C/C0) vs. time as shown in Fig. 5b. The apparent rate constants are determined to be 2.35, 4.7, 8.73 and 11.5 × 10−3 min−1 at the reaction temperatures of 30, 40 and 50 °C, respectively. Based on the Arrhenius plot (Fig. 5c), the activation energy of the degradation process is calculated to be 616 kJ mol−1.
 |
| | Fig. 5 (a) Effects of temperature in the degradation of CR, (b) plotting of ln(C/C0) vs. time and (c) Arrhenius plot of lnk vs. 1/T. Experimental conditions: catalyst loading = 100 mg L−1; initial CR concentration = 100 mg L−1; H2O2 concentration = 3 wt%; pH = 7. | |
To study the pathway of CR degradation, the degradation products were analyzed by HPLC-MS. Six products are identified as shown in Fig. S1† and the evolution of the products is illustrated in Fig. 6. It is found that the asymmetrical cleavage of C–N bonds is the first step of CR degradation. Subsequently, the resulting compound #4 is degraded via four different routes, leading to the generation of compounds #3, #1, #2, #5 and #6 as shown in Fig. 6. The degradation products obtained in this work were consistent with those reported in the literature.50,51 The symmetrical cleavage between the two benzene rings in CR molecules was reported to be a second pathway of CR degradation.52 Notably, such a degradation pathway of CR does not occur in the Co0.5Cu0.5O/H2O2 system.
 |
| | Fig. 6 Proposed CR degradation pathways in the Co0.5Cu0.5O-catalyzed AOP process. | |
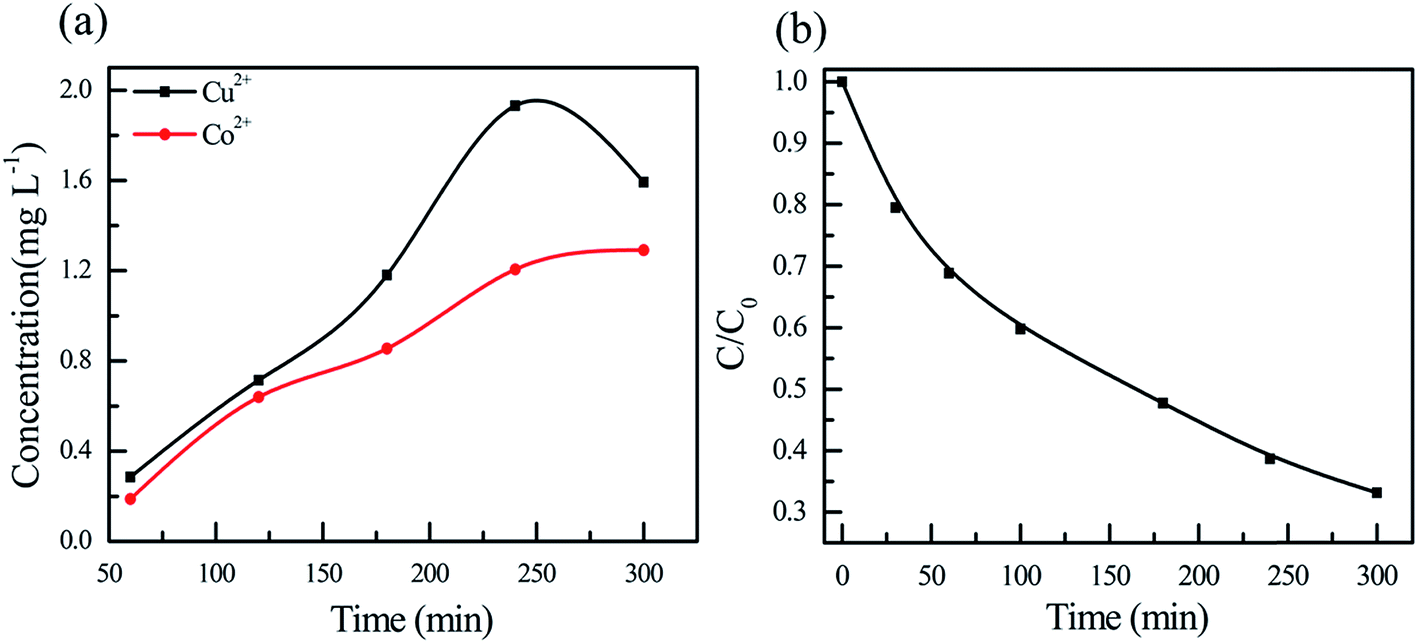
The metal leaching is a critical issue of many heterogeneous Fenton catalysts for practical application. The concentrations of cobalt and copper ions in the solution were monitored as shown in Fig. 7a. The concentration of cobalt ions increases with reaction time while the concentration of copper first increases with increasing time from 60 to 240 min but decreases with further increasing time to 300 min, yielding a maximum concentration at 240 min. The concentration of cobalt is always lower than that of copper. It is noted that the maximum of copper ion concentration is 1.9 mg L−1, which is lower than the emission standard of 2 mg L−1.27 The variation of H2O2 concentration in the pollutant solution was determined as a function of time. As shown in Fig. 7b, the H2O2 concentration decreases with increasing reaction time. Careful inspection reveals that the concentration of H2O2 showed the similar trend with that of CR, confirming that the degradation of CR pollutant is directly related to the activation of H2O2. The constant pH values of the solution are also monitored as shown in Fig. S2.† With an initial pH of 5, the constant pH monotonously increases with increasing reaction time. Interestingly, when the solutions with initial pHs of 7, 9 and 11 are used, the constant pH solution first decreases in the initial periods of reaction time and then increases with increasing time. The variations of pH are probably related to the degradation products in the degradation process.
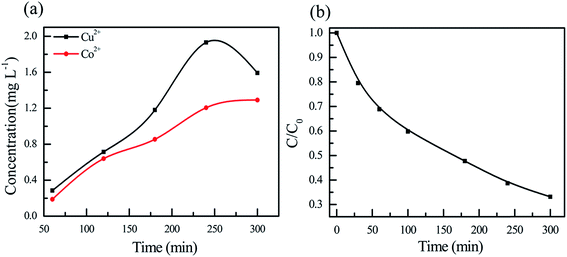 |
| | Fig. 7 (a) Variations of (a) metal ion and (b) H2O2 concentrations as a function of time. | |
The role of heterogeneous catalysts in the Fenton process is to activate H2O2 molecules to produce oxidative oxygen-containing species. The activation mechanism of H2O2 in the cobalt oxide catalyst is attributed to the redox pairs of CoII/CoIII as described by eqn (3)–(6):28
| | | H2O2 + OH− ⇔ OOH− + H2O | (3) |
| | ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CoII + H2O2 → CoIII + ˙OH + OH− CoII + H2O2 → CoIII + ˙OH + OH− | (4) |
| | | CoIII + OOH− → CoII˙OOH | (5) |
| | | CoII˙OOH → CoII + ˙O2H | (6) |
It was demonstrated that the Co0.5Cu0.5O catalyst showed superior catalytic performance at a pH value of 9 in comparison with the pH values of 5, 7 and 11 (see Fig. 4c). This can be well interpreted from the above mechanism. At high pH values, a larger number of perhydroxyl anions (OOH−) are available in the solution viaeqn (3). Compared with H2O2 molecules, OOH− is more nucleophilic and show higher activity to interact with CoIII in the Co0.5Cu0.5O catalyst (eqn (5)).28 Therefore, a high pH value in the pollutant solution facilitates the activation of H2O2 to generate oxidative oxygen-containing species, thus accelerating the degradation of CR pollutant. However, in strong alkaline solutions, (e.g. pH = 11), H2O2 becomes unstable and decomposes into water and oxygen (eqn (7)),53 which significantly decreases H2O2 concentration but without gain of oxidative radicals. Therefore, the degradation performance of the Co0.5Cu0.5O catalyst is reduced significantly at pHs over 9.
In the Co0.5Cu0.5O catalyst, the copper ions also play an important role on the degradation of CR pollutant. On one hand, the redox pairs CuII/CuI can activate H2O2 molecules to produce radicals based on eqn (8) and (9):26
| | | CuII + H2O2 → CuI + ˙O2− + 2H+ | (8) |
| | | CuI + H+ + H2O2 → CuII + OH + H2O | (9) |
The superior catalytic activity of binary metal oxides could also be explained from the critical role of CuI, which showed outstanding ability to degrade azo dyes via the Sandmeyer equation.27 In this study, the resulting CuI is capable of breaking the –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– bonds via the Sandmeyer reaction to generate carbon radicals. Compared with CR molecules, these carbon intermediates would react with radicals more easily, resulting in faster degradation.
N– bonds via the Sandmeyer reaction to generate carbon radicals. Compared with CR molecules, these carbon intermediates would react with radicals more easily, resulting in faster degradation.
| | | Azo compounds + CuI → carbon center radicals + CuII | (10) |
In addition, it should be pointed out that the presence of copper could also facilitate the reduction of CoIII. Considering the standard reduction potentials of E0 (CoII/CoIII) = 1.81 V and E0 (CuII/CuI) = 0.15 V, the reduction of CoIII by CuI would be thermodynamically favorable, as shown in eqn (11):29
| | | CoIII + CuI → CoII + CuII | (11) |
Therefore, the efficient regeneration of CoII and CuII in the catalyst surface by this process would also be responsible for the superior catalytic activity of the Co0.5Cu0.5O catalyst.
To verify the above-mentioned mechanism, tert-butanol and chloroform were separately added into the pollutant solution as scavengers for hydroxyl radical and superoxide radical, respectively.54 Three different molar ratios of scavengers and CR (10:1, 50:1, and 100:1) were used for the parallel experiments and the blank test without scavenger was also included for comparison. Fig. 8 shows the effects of t-butanol and chloroform. It is indicated that the presence of t-butanol and chloroform in the solutions significantly depresses the degradation of CR. These results clearly indicate that both hydroxyl radical and superoxide radical are generated during the H2O2 activation, which is well consistent with the aforementioned mechanisms.
 |
| | Fig. 8 Effects of tert-butanol (a) and chloroform in degradation rate of CR. Experimental conditions: catalyst loading = 100 mg L−1; initial CR concentration = 100 mg L−1; pH = 7; H2O2 concentration = 3 wt%; temperature = 30 ± 1 °C. | |
The recyclability of the Co0.5Cu0.5O catalyst was examined. The Co0.5Cu0.5O catalyst was tested in seven successive runs to study its stability and regenerability. In each cycle, the catalyst was tested for 60 min, separated by centrifugation, and then regenerated via combustion at 400 °C in air for 2 h. Fig. 9 displays the CR degradation of different cycles. It is indicated that after removing surface contaminants, the Co0.5Cu0.5O catalyst recovers its excellent activity. The activity of Co0.5Cu0.5O catalyst shows very limited decay due to the loss of catalyst during the separation and regeneration processes. Overall, although the efficiency of Co0.5Cu0.5O mediated Fenton process reported in this work is slightly inferior to that of homogeneous Fenton process reported by Ay et al.,55 the Co0.5Cu0.5O catalyst displayed better efficiency as compared with the biological processes,56,57 and the CuO nanosheets.58
 |
| | Fig. 9 Cyclic tests of Co0.5Cu0.5O catalyst. Experimental conditions: catalyst loading = 200 mg L−1; CR concentration = 100 mg L−1; pH = 9; H2O2 concentration = 3 wt%; temperature = 30 ± 1 °C. | |
Conclusions
CoO, CuO, Co0.75Cu0.25O and Co0.5Cu0.5O were obtained by simple calcination of corresponding oxalates. The oxides were composed of fine nanoparticles with high crystallinity. XRD results confirmed the formation of solid solutions in the binary metal oxides. The resulting oxides were used as heterogeneous catalysts for the Fenton-degradation of CR. Among the four catalysts, the Co0.5Cu0.5O catalyst showed the best catalytic performance. The degradation of CR is highly dependent on the substrate concentration, pH, H2O2 concentration and reaction temperature. In contrast with the homogeneous Fenton reaction, which requires strong acidic pHs of the pollutant solutions, the heterogeneous Fenton reaction mediated by the Co0.5Cu0.5O showed outstanding activity at a pH value of 9. It was found that the optimal H2O2 concentration of 4% facilitated the degradation reaction and that the degradation performance increased with increasing the reaction temperature. The degradation products were identified and the CR degradation was initiated by the asymmetric cleavage of C–N bonds in CR molecules. Further mechanistic studies revealed that hydroxyl and superoxide radicals were mainly responsible for the degradation of CR. The Co0.5Cu0.5O catalyst showed the best catalytic performance compared with the three other oxides, which was attributed to the critical role of copper ions. The metal leaching of the Co0.5Cu0.5O catalyst was quite limited. The Co0.5Cu0.5O catalyst could be facilely regenerated by combustion and showed excellent recyclability. Overall, the Co0.5Cu0.5O/H2O2 system showed promising application for dye degradation in aqueous solutions.
Acknowledgements
The Project was financially supported by the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry and the Natural Science Foundation of Guangdong Province, China (2014A030310315).
References
- T. Robinson, G. McMullan, R. Marchant and P. Nigam, Bioresour. Technol., 2001, 77, 247–255 CrossRef CAS.
- N. Azbar, T. Yonar and K. Kestioglu, Chemosphere, 2004, 55, 35–43 CrossRef CAS PubMed.
- E. Forgacs, T. Cserhátia and G. Oros, Environ. Int., 2004, 30, 953–971 CrossRef CAS PubMed.
- H. J. H. Fenton, J. Chem. Soc., Trans., 1894, 65, 899–910 RSC.
-
(a) E. Ember, H. A. Gazzaz, S. Rothbart, R. Puchta and R. van Eldik, Appl. Catal., B, 2010, 95, 179–191 CrossRef CAS PubMed;
(b) I. Popivker, I. Zilbermann, E. Maimon, H. Cohen and D. Meyerstein, Dalton Trans., 2013, 42, 16666–16668 RSC.
- P. V. Nidheesh, RSC Adv., 2015, 5, 40552–40577 RSC.
- M. Klavarioti, D. Mantzavinos and D. Kassinos, Environ. Int., 2009, 35, 402–417 CrossRef CAS PubMed.
- X. N. Li, J. H. Wang, A. I. Rykov, V. K. Sharma, H. Z. Wei, C. Z. Jin, X. Liu, M. R. Li, S. H. Yu, C. L. Sun and D. D. Dionysioud, Catal. Sci. Technol., 2015, 5, 504–514 CAS.
- S. Chakma and V. S. Moholkar, RSC Adv., 2015, 5, 53529–53542 RSC.
- W. P. Liu, L. L. Xu, X. F. Li, C. S. Shen, S. Rashid, Y. Z. Wen, W. P. Liu and X. H. Wu, RSC Adv., 2015, 5, 2449–2456 RSC.
- H. Wang, H. Jiang, S. Wang, W. B. Shi, J. C. He, H. Liu and Y. M. Huang, RSC Adv., 2014, 4, 45809–45815 RSC.
- L. Yu, X. F. Yang, Y. S. Ye and D. S. Wang, RSC Adv., 2015, 5, 46059–46066 RSC.
- S. Kundu, A. Chanda, J. V. K. Thompson, G. Diabes, S. K. Khetan, A. D. Ryabov and T. J. Collins, Catal. Sci. Technol., 2015, 5, 1775–1782 CAS.
- J. Wang, C. Liu, L. Tong, J. S. Li, R. Luo, J. W. Qi, Y. Li and L. J. Wang, RSC Adv., 2015, 5, 69593–69605 RSC.
- Z.-T. Hu, B. Chen and T.-T. Lim, RSC Adv., 2014, 4, 27820–27829 RSC.
- S. Kouraichi, M. El-Hadi Samar, M. Abbessi, H. Boudouh and A. Balaska, Catal. Sci. Technol., 2015, 5, 1052–1064 Search PubMed.
- A. Dhakshinamoorthy, S. Navalon, M. Alvaro and H. Gacia, ChemSusChem, 2012, 5, 46–64 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, S. Navalon, M. Alvaro and H. Gacia, ChemSusChem, 2011, 4, 1712–1730 CrossRef PubMed.
- S. Navalon, M. Alvaro and H. Garcia, Appl. Catal., B, 2010, 99, 1–26 CrossRef CAS PubMed.
- J. Herney-Ramirez, M. A. Vicente and L. M. Madeira, Appl. Catal., B, 2010, 98, 10–26 CrossRef CAS PubMed.
- I. S. X. Pinto, P. H. V. V. Pacheco, J. V. Coelho, E. Lorencon, J. D. Ardisson, J. D. Fabris, P. P. de Souza, K. W. H. Krambrock, L. C. A. Oliveira and M. C. Pereira, Appl. Catal., B, 2012, 119–120, 175–182 CrossRef CAS PubMed.
- C. M. Zheng, X. Z. Cheng, P. P. Chen, C. W. Yang, S. M. Bao, J. Xia, M. L. Guo and X. H. Sun, RSC Adv., 2015, 5, 40872–40883 RSC.
- L. C. Zhou, H. Zhang, L. Q. Ji, Y. M. Shao and Y. F. Li, RSC Adv., 2014, 4, 24900–24908 RSC.
- J. G. Liu, C. J. Ou, W. Q. Han, Faheem, J. Y. Shen, H. P. Bi, X. Y. Sun, J. S. Li and L. J. Wang, RSC Adv., 2015, 5, 57444–57452 RSC.
- M. Munoz, Z. M. de Pedro, J. A. Casas and J. J. Rodriguez, Appl. Catal., B, 2015, 176, 249–265 CrossRef PubMed.
- B. J. Dramou, V. Shah and J. M. Pinto, Energy Environ. Sci., 2008, 1, 395–402 CAS.
- G. H. Dong, Z. H. Ai and L. Z. Zhang, Water Res., 2014, 66, 22–30 CrossRef CAS PubMed.
- J. S. Ma, L. Zhang, M. Zhao and Y. Wang, J. Mol. Catal. A: Chem., 2013, 378, 30–37 CrossRef PubMed.
- R. C. C. Costa, M. F. F. Lelis, L. C. A. Oliveira, J. D. Fabris, J. D. Ardisson, R. R. V. A. Rios, C. N. Silva and R. M. Lago, J. Hazard. Mater., 2006, 129, 171–178 CrossRef CAS PubMed.
- C. Hu, S. T. Xing, J. H. Qu and H. He, J. Phys. Chem. C, 2008, 112, 5978–5983 CAS.
- T. Rhadfi, J. Y. Piquemal, L. Sicard, F. Herbst, E. Briot, M. Benedetti and A. Atlamsani, Appl. Catal., A, 2010, 386, 132–139 CrossRef CAS PubMed.
- S. H. Do, B. Batchelor, H. K. Lee and S. H. Kong, Chemosphere, 2009, 75, 8–12 CrossRef CAS PubMed.
- A. C. Lua and H. Y. Wang, Appl. Catal., B, 2013, 132–133, 469–478 CrossRef CAS PubMed.
- G. M. Eisenberg, Ind. Eng. Chem., Anal. Ed., 1943, 15, 327–328 CrossRef CAS.
- W. Luo, L. H. Zhu, N. Wang, H. Q. Tang, M. J. Cao and Y. B. She, Environ. Sci. Technol., 2010, 44, 1786–1791 CrossRef CAS PubMed.
- X. L. Liang, Y. H. Zhong, S. Y. Zhu, J. X. Zhu, P. Yuan, H. P. He and J. Zhang, J. Hazard. Mater., 2010, 181, 112–120 CrossRef CAS PubMed.
- C. G. Silva and J. L. Faria, ChemSusChem, 2010, 3, 609–618 CrossRef CAS PubMed.
- A. L. T. Pham, F. M. Doyle and D. L. Sedlak, Water Res., 2012, 46, 6454–6462 CrossRef CAS PubMed.
- Q. J. Yang, H. Choi, S. R. Al-Abed and D. D. Dionysiou, Appl. Catal., B, 2009, 88, 462–469 CrossRef CAS PubMed.
- Q. Wang, S. L. Tian and P. Ning, Ind. Eng. Chem. Res., 2014, 53, 6334–6340 CrossRef CAS.
- C. Liu, J. S. Li, J. W. Qi, J. Wang, R. Luo, J. Y. Shen, X. Y. Sun, W. Q. Han and L. J. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 13167–13173 CAS.
- L. J. Xu and J. L. Wang, Environ. Sci. Technol., 2012, 46, 10145–10153 CAS.
- M. F. Variava, T. L. Church and A. T. Harris, Appl. Catal., B, 2012, 123–124, 200–207 CrossRef CAS PubMed.
- J. A. Melero, G. Calleja, F. Martinez, R. Molina and M. I. Pariente, Chem. Eng. J., 2007, 131, 245–256 CrossRef CAS PubMed.
- M. L. Luo, D. Bowden and P. Brimblecombe, Appl. Catal., B, 2009, 85, 201–206 CrossRef CAS PubMed.
- Y. L. Zhang, K. Zhang, C. M. Dai, X. F. Zhou and H. P. Si, Chem. Eng. J., 2014, 244, 438–445 CrossRef CAS PubMed.
- J. Fernández, J. Kiwi, J. Baeza, J. Freer, C. Lizama and H. D. Mansilla, Appl. Catal., B, 2004, 48, 205–211 CrossRef PubMed.
- G. B. O. de la Plata, B. Guadalupe, O. M. Alfano and A. E. Cassano, Appl. Catal., B, 2010, 95, 1–13 CrossRef PubMed.
- S. H. Tian, Y. T. Tu, D. S. Chen, X. Chen and Y. Xiong, Chem. Eng. J., 2011, 169, 31–37 CrossRef CAS PubMed.
- Z. X. Chen, D. Z. Li, W. J. Zhang, Y. Shao, T. W. Chen, M. Sun and X. Z. Fu, J. Phys. Chem. C, 2009, 113, 4433–4440 CAS.
- Y. Yang, Q. Y. Wu, Y. H. Guo, C. W. Hu and E. B. Wang, J. Mol. Catal. A: Chem., 2005, 225, 203–212 CrossRef CAS PubMed.
- X. L. Li, J. Zhang, Y. C. Jiang, M. C. Hu, S. N. Li and Q. G. Zhai, Ind. Eng. Chem. Res., 2013, 52, 13572–13579 CrossRef CAS.
- W. Chu, W. K. Choy and T. Y. So, J. Hazard. Mater., 2007, 141, 86–91 CrossRef CAS PubMed.
- S. Bae, D. Kim and W. Lee, Appl. Catal., B, 2013, 134–135, 93–102 CrossRef CAS PubMed.
- F. Ay, E. C. Catalkaya and F. Kargi, J. Hazard. Mater., 2009, 162, 230–236 CrossRef CAS PubMed.
- A. A. Telke, S. M. Joshi, S. U. Jadhav, D. P. Tamboli and S. P. Govindwar, Biodegradation, 2010, 21, 283–296 CrossRef CAS PubMed.
- O. D. Olukanni, A. A. Osuntoki, A. O. Awotula, D. C. Kalyani, G. O. Gbenle and S. P. Govindwar, J. Microbiol. Biotechnol., 2013, 236, 843–849 CrossRef PubMed.
- A. Sadollahkhani, Z. H. Ibupoto, S. Elhag, O. Nur and M. Willander, Ceram. Int., 2014, 40, 11311–11317 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra18923c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.