
Two-step process for programmable removal of oxygen functionalities of graphene oxide: functional, structural and electrical characteristics†
Abstract
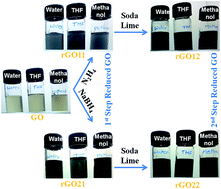
Here we report a two-step programmable reduction of graphene oxide (GO) which was synthesized by oxidation of graphite. X-Ray photoelectron spectroscopic (XPS) analysis confirmed the synthesis of exfoliated graphene oxide (GO) by introduction of oxygen as carboxylic (–COOH), epoxy (C–O–C) and hydroxyl (–OH) groups. The first step of GO reduction was achieved separately by (i) hydrazine (rGO11) and (ii) sodium borohydride (rGO21). Soda lime was used in the second-stage reduction of (a) hydrazine reduced GO (rGO12) and (b) sodium borohydride reduced GO (rGO22) to remove most of the remaining carboxylic functionalities from the rGO11 and rGO21 surface. XPS spectra of rGO21 showed a decrease (38 to 30%) in the oxygen whereas the further reduction of rGO21 with soda lime can further reduce the oxygen content. Quantitative analysis of C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)OX in GO shows about 43% of carbon atoms (C 1s signal) as carboxylic functionalities whereas the reduction of the GO with sodium borohydride reduced this signal to about 10%. The use of soda lime for both rGO11 and rGO21 further reduced the amount of carboxylic functionalities. An increase in the proportion of carbon atoms as sp2 and decrease in the oxygen functionalities were controlled in the two-step reduction. A good correlation in the conductivity of reduced GO with the percentage proportion of sp2 carbon was observed.
O)OX in GO shows about 43% of carbon atoms (C 1s signal) as carboxylic functionalities whereas the reduction of the GO with sodium borohydride reduced this signal to about 10%. The use of soda lime for both rGO11 and rGO21 further reduced the amount of carboxylic functionalities. An increase in the proportion of carbon atoms as sp2 and decrease in the oxygen functionalities were controlled in the two-step reduction. A good correlation in the conductivity of reduced GO with the percentage proportion of sp2 carbon was observed.
 Please wait while we load your content...
Please wait while we load your content...

