
Synthesis and aggregation-induced emission properties of pyridine and pyridinium analogues of tetraphenylethylene†
Abstract
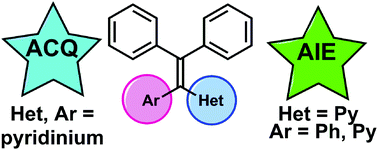
Aggregation-induced emission (AIE) is emerging as an important design element in a variety of new fluorescent-based chemical sensors and bio-imaging agents. In particular, derivatives of tetraphenylethylene (TPE) have been widely utilized in this regard as the TPE framework is a reliable AIE-luminogen. To expand the library of AIE active tetraarylethylenes, we have explored the effects of replacing one or two of the phenyl rings in TPE with pyridine. Efficient synthetic routes that deliver mono- and bis-pyridyl tetraarylethylenes have been developed and the luminescent properties of these heterocyclic TPE analogues, along with their corresponding N-methylated pyridinium salts, have been examined.
 Please wait while we load your content...
Please wait while we load your content...

