
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe promotional role of Ce in Cu/ZSM-5 and in situ surface reaction for selective catalytic reduction of NOx with NH3†
Shuangshuang
Lai
,
Dongmei
Meng
,
Wangcheng
Zhan
*,
Yun
Guo
,
Yanglong
Guo
,
Zhigang
Zhang
and
Guanzhong
Lu
*
Key Laboratories for Advanced Materials, Research Institute of Industrial Catalysis, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China. E-mail: gzhlu@ecust.edu.cn; zhanwc@ecust.edu.cn; Fax: +86-21-64252923
First published on 12th October 2015
Abstract
Cu/ZSM-5 and Ce doped Cu/ZSM-5 catalysts were prepared by the incipient-wetness-impregnation method, and the effect of Ce doping on the structure and the catalytic performance of the Cu/ZSM-5 catalyst was investigated in detail for the selective catalytic reduction (SCR) of NO with NH3. The results showed that the addition of Ce can markedly broaden the operation temperature window of the Cu/ZSM-5 catalyst for NH3-SCR and enhance its H2O and SO2 resistance. The presence of Ce promoted an enrichment of copper species (isolated Cu2+ ions and CuO nanoparticles) on the catalyst surface and the increase in the Lewis acid sites on the surface of the Cu/ZSM-5 catalyst, and strengthened the redox property of the Cu/ZSM-5 catalyst. As a result, Ce-doped Cu/ZSM-5 catalyst possessed the high adsorption ability of NH3 and nitrite/nitrate, which is propitious to the increase in the reactivity of the Ce-doped Cu/ZSM-5 catalyst. In situ DRIFTS results indicated that the NH3-SCR reaction on the Cu/ZSM-5 catalyst and Ce1–Cu4/ZSM-5 catalysts definitely followed Langmuir–Hinshelwood mechanisms, and bridged nitrates and bidentate nitrates were the active intermediate. However, Eley–Rideal mechanism can't be confirmed over the Cu/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts.
1. Introduction
Nitrogen oxides (NOx) originated from various combustion processes are major air pollutants, which are very harmful to human health, due to the formation of photochemical smog, acid rain, ozone depletion and greenhouse effects. The selective catalytic reduction of NOx with ammonia (NH3-SCR) in the presence of excess oxygen has been considered as one of the best available approaches to control the emission of NOx produced from the stationary sources and the diesel vehicle. V2O5–WO3(MoO3)/TiO2 behaves the high activity and selectivity of NH3-SCR and has been the most widely used in industry.1 However, there are still some inevitable problems, such as the narrow working temperature window (300–400 °C), the toxicity of vanadium species,2 and the low N2 selectivity at high temperatures.3 Therefore, it is necessary to develop vanadium-free catalysts with high SCR activity.In recent years, the zeolite-based catalysts are considered to be a practical solution for reducing the exhaust pollutants from diesel engines, and exhibit the high activities and good stability at high temperatures,4,5 especially the Cu/ZSM-5 catalyst due to its superior SCR activity and N2 selectivity in a wide temperature range.6 However, previous researches indicated that the Cu/ZSM-5 catalyst did not possess a high SCR activity at high temperature.7 With respect to monometallic copper loaded catalysts, it is difficult to enhance its SCR activity simply by increasing the copper content in the Cu/ZSM-5 catalyst. This is because a high content of copper inevitably leads to agglomeration of the copper species and formation of large copper oxide particles,8 which would enhance the oxidation of NH3 and narrow the temperature window. It has been determined that the optimal content of copper in the Cu/ZSM-5 catalyst is within 3–4 wt%.9
Ceria has been studied extensively as an oxygen reservoir, which can store and release oxygen by the redox cycle between Ce3+ and Ce4+ under oxidizing and reducing conditions.10–12 Many Ce doped catalysts were studied extensively and used in the various reactions, including CeO2–MnOx,10,13 CeWOx,14 Ce–Mn/ZSM-5,11 and Ce–Cu/ZSM-5.15,16 Recently, it was reported that Ce–Cu/ZSM-5 catalyst was used for NH3-SCR reaction. Pang et al.17 reported that adding Ce into Cu/ZSM-5 monolithic catalyst could obviously improve its activity and hydrothermal stability by stabilizing the dispersion of CuO and suppressing the formation of bulk-type CuO crystallites during hydrothermal treatment. Dou et al.18 also confirmed that Ce doping improved the redox properties of the Cu–Ce/ZSM-5 catalyst, due to the higher valence of copper and mobility of lattice oxygen than those of Cu/ZSM-5 catalyst. But little attention has been paid to the effect of Ce doping on the acid sites, surface species and adsorption property of Ce–Cu/ZSM-5 catalysts. And the surface reaction mechanism over the Ce–Cu/ZSM-5 catalyst for the NH3-SCR reaction and the role of Ce on the catalytic cycle has not been investigated.
Herein, we try to solve two problems for Ce doped Cu/ZSM-5 catalysts, the promotional role of Ce and the surface reaction mechanism over the Ce–Cu/ZSM-5 catalyst for the NH3-SCR reaction, including the effect of Ce doping on the SO2 resistance and water resistance. These research results will certainly be useful for the further development of the zeolite catalysts for the NH3-SCR reaction.
2. Experimental
2.1 Preparation of catalysts
A series of Cex–Cu4/ZSM-5 catalysts with copper content of 4 wt% and variable content of cerium were prepared by an improved incipient-wetness-impregnation method. H–ZSM-5 with Si/Al atomic ratio of 18 was supplied by Nankai University, Tianjin, China. In a typical process, the required amount of an aqueous solution of Cu(NO3)2·3H2O and Ce(NO3)3·6H2O was slowly dropped into the support under vigorous stirring at room temperature, and then ultrasonically treated for 1 h. The content of copper in the catalysts was maintained at 4 wt%, and the content of cerium was varied. The solid was dried at 120 °C for 12 h and calcined in air at 550 °C for 4 h. The final catalysts were labeled as Cex–Cu4/ZSM-5 (x denotes the weight ratio of Ce/H–ZSM-5, x = 0.5, 1, 2 wt%).The Cu4/ZSM-5 catalyst was also prepared with the same procedure as Cex–Cu4/ZSM-5, but no Ce(NO3)3·6H2O was added in the synthesis solution.
2.2 Catalyst characterization
The XRD patterns were recorded on a Brook/D8 diffractometer with CuKα Radiation (λ = 0.154056 nm) in the 2θ range of 5–60°. The morphologies of the catalysts were investigated by field emission scanning electron microscopy (FE-SEM) on a Hitachi S-4800 instrument operated at the beam energy of 15 kV. The N2 adsorption–desorption isotherms were measured on a Quantachrome NOVA1200 surface area at −196 °C. Prior to the measurements, all samples were degassed at 180 °C until a stable vacuum of ca. 5 mTorr was reached. The surface areas of the samples were calculated by Brunauer–Emmett–Teller (BET) method. The pore size distribution were calculated from the desorption branch using the Barrett–Joyner–Halenda (BJH) method. The XPS spectra were recorded on a Thermo ESCALAB 250 spectrometer with a monochromatized AlKα X-ray source (1486.6 eV) and a passing energy of 25 eV. C1s (binding energy 284.6 eV) of adventitious carbon was used as the reference.Py-IR spectra of samples were analyzed by a Nicolet 5700 FT-IR spectrometer. The samples (13 mg) was heated at 400 °C under vacuum for 2 h, and cooled to 200 °C when pyridine was chemisorbed for 10 min. After this step, the sample was evacuated and analyzed by FTIR.
Temperature-programmed desorption of NH3 (NH3-TPD) was carried out on a PX200 apparatus (Tianjin Pengxiang Technology Ltd. China) with a thermal conductivity detector (TCD). 50 mg of the sample was filled into the quartz reactor and pretreated at 500 °C in a flow of N2 (50 mL min−1) for 1 h. After being cooled down to room temperature, the sample was exposed to a flow of 10% NH3/N2 (50 mL min−1) for 0.5 h. Then the sample was flushed by N2 (50 mL min−1) for 1 h and NH3-TPD was carried out by heating the sample in N2 (50 mL min−1) from room temperature to 500 °C at 10 °C min−1.
NO-TPD was carried out on the custom-made equipment with a NOx analyzer (Thermo Fisher Model 42i-HL NO–NOx-chemiluminescence analyzer) as detector. The sample was pretreated in Ar (450 mL min−1) at 450 °C for 1 h and cooled down to room temperature. Then the sample was exposed to a flow of 500 ppm NO/Ar (300 mL min−1) for 1 h to reach saturated adsorption of NO on the sample, followed by Ar (300 mL min−1) purging for 1 h. Finally, NO-TPD was carried out by heating the sample in Ar (300 mL min−1) from room temperature to 450 °C at 10 °C min−1.
Temperature-programmed reduction with H2 (H2-TPR) was performed in a conventional flow apparatus. 100 mg of the sample was used. A flow of 5% H2/N2 (40 mL min−1) was passed through the catalyst bed at 10 °C min−1 from 30 to 600 °C. The consumption of hydrogen was monitored by a thermal conductivity detector (TCD).
The in situ DRIFT measurements were performed on a Nicolet 6700 FT-IR spectrometer with a MCT detector. The sample was pretreated at 500 °C in Ar for 1 h, and then cooled to 150 °C in Ar. The background spectra were recorded at the different temperatures during the cooling, and background was subtracted from sample spectra accordingly.
2.3 Catalytic activity testing
The catalytic activities of the Cex–Cu4/ZSM-5 catalysts for NH3-SCR in excess oxygen were investigated in a fixed-bed quartz reactor (Φ 6 mm × 300 mm). 200 mg of the catalyst (20–40 mesh) was used. The reactant gas was composed of 500 ppm NO, 500 ppm NH3, 5 vol% O2, 2 vol% H2O (when used), 50 ppm SO2 (when used) and balanced Ar. The gas hourly space velocity (GHSV) was 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. The concentrations of NO and NO2 remained in the product were analysed by a Thermo Fisher NO–NOx-chemiluminescence analyzer. To avoid modest errors caused by the oxidation of ammonia in the converter of NO/NOx analyzer, an ammonia trap containing phosphoric acid solution was installed prior to the chemiluminescence detector. NOx conversion X(NOx) was calculated as follows:
000 h−1. The concentrations of NO and NO2 remained in the product were analysed by a Thermo Fisher NO–NOx-chemiluminescence analyzer. To avoid modest errors caused by the oxidation of ammonia in the converter of NO/NOx analyzer, an ammonia trap containing phosphoric acid solution was installed prior to the chemiluminescence detector. NOx conversion X(NOx) was calculated as follows:
3. Results and discussion
3.1 Catalytic performance for NH3-SCR activity
The catalytic activities of the Cu4/ZSM-5 and Cex–Cu4/ZSM-5 catalysts for the NH3-SCR reaction are shown in Fig. 1. The H–ZSM-5 catalyst exhibited a very low activity, over which the maximum NOx conversion is only 80% at 425 °C. The Cu4/ZSM-5 catalyst has a high catalytic activity, and the NOx conversion of above 90% was obtained at 195–435 °C.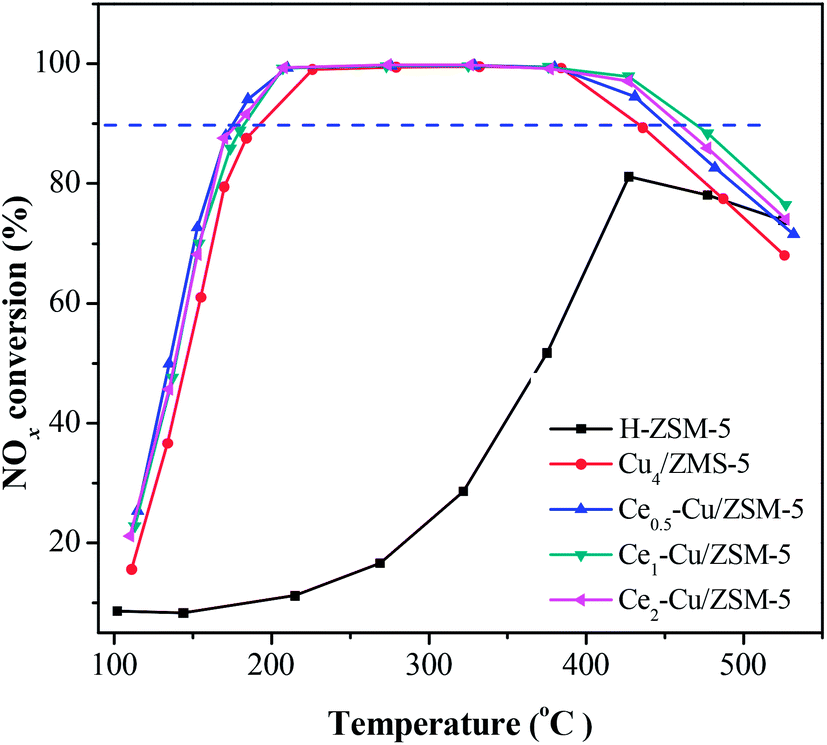 | ||
| Fig. 1 NOx conversion as a function of the reaction temperature over Cu/ZSM-5 and Cex–Cu4/ZSM-5 catalysts for the NH3-SCR reaction. (Reaction conditions: 0.2 g catalyst, the reactant gas of 500 ppm NO/500 ppm NH3/5% O2/Ar balanced, GHSV = 55000 h−1). | ||
When cerium was added to Cu4/ZSM-5, the catalytic activity of Cu4/ZSM-5 toward NH3-SCR reaction was significantly improved, especially at the high temperature. For example, when the Ce1–Cu4/ZSM-5 catalyst was used, the temperature window of more than 90% NOx conversion was 185–470 °C; while for the Ce2–Cu4/ZSM-5 and Ce0.5–Cu4/ZSM-5 catalysts, their window were 180–460 °C and 175–450 °C, respectively. The results clearly demonstrate that comparing with the operation window of Cu4/ZSM-5 catalyst, adding an appropriate amount of Ce in this catalyst can broaden the operation window about 45 °C. Furthermore, the amount of Ce obviously affects the NH3-SCR activity of the Cex–Cu4/ZSM-5 catalyst at the high temperature, while this influence is faint at the low temperature.
On the other hand, both the Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts behaved the high stability after the NH3-SCR reaction. As shown in Fig. S1,† the NH3-SCR activities of the Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts on the second run are almost the same as those on the first run.
3.2 Physicochemical properties of catalysts
Fig. 2 shows the XRD patterns of H–ZSM-5, Cu4/ZSM-5 and Cex–Cu4/ZSM-5 catalysts. All the samples exhibit the typical diffraction peaks of ZSM-5 zeolite at 2θ = 7.9°, 8.8°, 23.1° and 23.8°, which represent the (011), (020), (051) and (033) planes, respectively.19 Comparing with the XRD spectrum of H–ZSM-5, the XRD spectra of Cu4/ZSM-5 and Cex–Cu4/ZSM-5 catalysts were changed inconspicuously, which indicates that the structure of zeolite support remains intact after adding Cu and Ce. On the other hand, the diffraction peaks of CuO are not detected for all catalysts, showing that the copper species are well dispersed on the surface of the ZSM-5 support, or aggregated crystallites are too small to be detected by XRD. Similarly, the diffraction peaks of CeO2 are not also detected for the Cex–Cu4/ZSM-5 catalysts with low Ce content. However, with increasing the Ce content to 2.0 wt%, the very weak diffraction peak of CeO2 can be observed (2θ = 28.2°) for the Ce2–Cu4/ZSM-5 catalyst, indicating the formation of CeO2 crystallites on the surface of the Ce2–Cu4/ZSM-5 catalyst.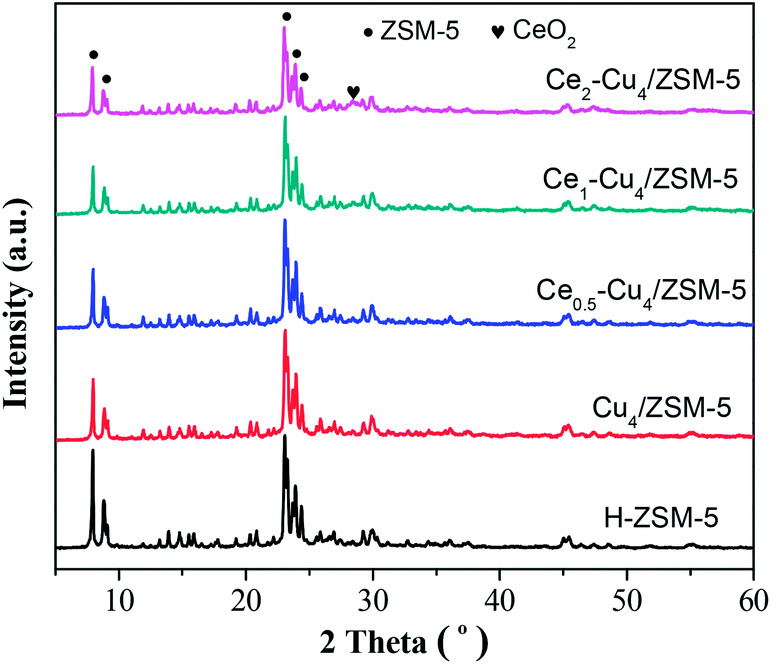 | ||
| Fig. 2 Wide-angle XRD patterns of pure H–ZSM-5, Cu4/ZSM-5 and Cex–Cu4/ZSM-5 catalysts. | ||
Fig. 3 shows the SEM images of H–ZSM-5, Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts. The results show that, H–ZSM-5 exhibits the schistose and irregular morphology, and after the addition of Cu and Ce, the morphologies of Cu4/ZSM-5 and Cex–Cu4/ZSM-5 catalysts are virtually unchanged. The BET surface area, average pore diameter and micro-pore volume are listed in Table 1. It can be seen that adding Cu leads to the decrease in BET surface area, average pore diameter and micro-pore volume, which can be attributed to the fact that copper species cover the external surface of H–ZSM-5, blocking many zeolite channels. After the addition of Ce into the Cu4/ZSM-5 catalyst, BET surface area was decreased slightly, while average pore diameter and micro-pore volume remained unchanged.
 | ||
| Fig. 3 SEM images of pure H–ZSM-5 (top left), Cu4/ZSM-5 (top right) and Ce1–Cu4/ZSM-5 (bottom). | ||
| Sample | S BET (m2 g−1) | Average pore diameter (nm) | Micro-pore volume (cm3 g−1) | Isolated Cu2+/CuOa | Surface composition (at%) | ||
|---|---|---|---|---|---|---|---|
| Cu (isolated Cu2+/CuO)b | Ce | O | |||||
| a The peak area ratio (m) of M (isolated Cu2+ ions)/M (CuO crystallites) in XPS spectrum. b “Isolated Cu2+” was calculated by Cu (at%) × (m/(1 + m)) and “CuO” was calculated by Cu (at%)/(1 + m). | |||||||
| H–ZSM-5 | 333 | 2.1 | 0.14 | — | — | — | — |
| Cu4/ZSM-5 | 280 | 1.6 | 0.12 | 0.239 | 1.59 (0.31/1.28) | — | 63.2 |
| Ce1–Cu4/ZSM-5 | 268 | 1.6 | 0.12 | 0.247 | 1.97 (0.39/1.58) | 0.25 | 62.8 |
Fig. 4 shows XPS Cu2p spectra of Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts. The Cu4/ZSM-5 catalyst has two main peaks at BE = 933.6 eV and 953.0 eV in its XPS Cu2p spectra, which are attributed to Cu2p3/2 and Cu2p1/2, respectively. The Cu2p3/2 peaks can be deconvoluted to two peaks, and the peaks at 933.4 eV represent the agglomerated CuO nanoparticles on the surface of catalysts, and the one at BE = 935.2 eV is attributed to isolated Cu2+ ion coordinated to superficial oxygen atoms of zeolite.18,20 Since the diffraction peaks of CuO species could not be observed in the XRD spectra, these CuO crystallites' size should be lower than 3 nm.
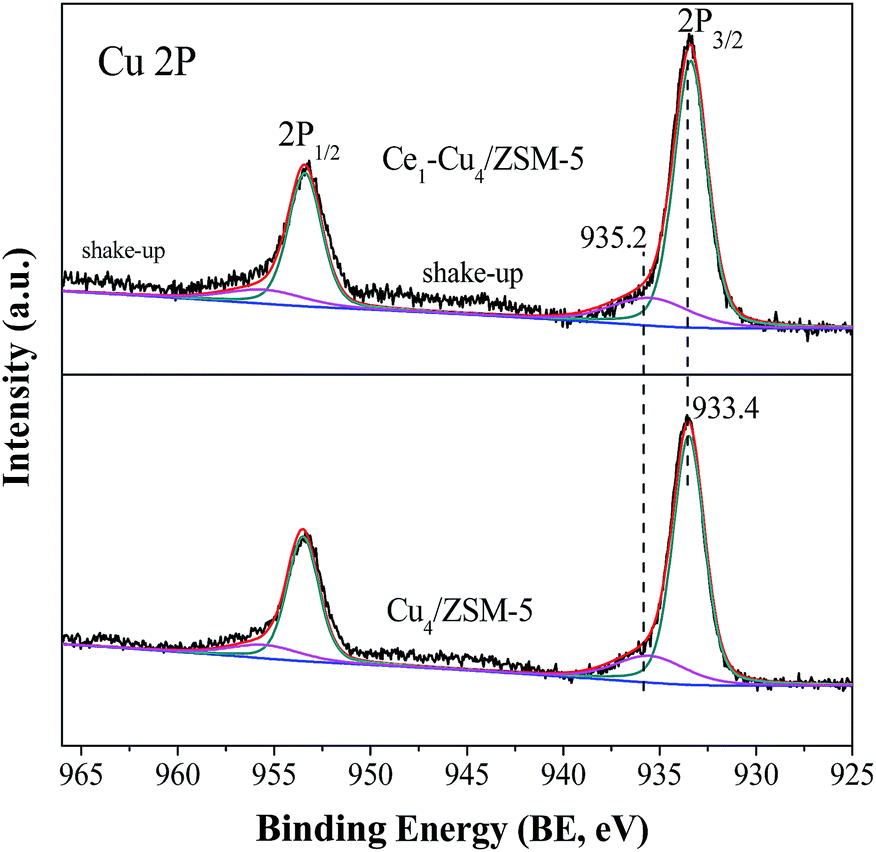 | ||
| Fig. 4 XPS Cu2p spectra of Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts. | ||
Table 1 shows the surface atom concentrations of Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts. It can be seen that surface Cu amount increases with the addition of Ce in the Cu4/ZSM-5 catalyst, indicating that the presence of Ce can help to the enrichment of Cu on the Cu4/ZSM-5 surface. According to the peak area ratio of (isolated Cu2+ ions)/(CuO crystallites) in XPS spectra, the amounts of isolated Cu2+ ions and CuO crystallites on the surface of Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts were calculated (Table 1). The results show that CuO crystallites are predominant on the surface of both Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts with a high Cu content, which is consistent with the reported results.20 Furthermore, the surface concentrations of isolated Cu2+ ions and CuO crystallites over the Ce1–Cu4/ZSM-5 catalyst are higher than that over the Cu4/ZSM-5 catalyst, because of the increase of the surface Cu atoms after Ce addition in the Cu4/ZSM-5 catalyst.
Wang et al.21 thought both CuO nanoparticles and isolated Cu2+ ion in Cu/SAPO-34 were active sites for the NH3-SCR reaction. Furthermore, it is revealed that the CuO nanoparticles catalyzes the oxidation of NO to NO2, and this favors the reduction of NO at lower temperature due to the facilitation of the “fast SCR” process, while isolated Cu2+ ions help to the high NO conversion at high temperature. Therefore, when adding Ce in the Cu4/ZSM-5 catalyst, the increase in the surface concentrations of both isolated Cu2+ ions and CuO nanoparticles can improve the activity of the Cu4/ZSM-5 catalyst.
Fig. 5 shows the UV-Vis absorption spectra of H-ZSM-5, Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts. H-ZSM-5 exhibits the adsorption bands at about 210 and 280 nm. For Cu4/ZSM-5, a band at 258 nm and a broad band centered at 750 nm between 550 and 800 nm appeared. The former can be assigned to O → Cu transitions of isolated Cu2+ ions in coordination with lattice oxygen, and the latter can be assigned to d–d transitions of Cu2+ ions in a octahedrically coordinated environment due to dispersed CuO crystallites on the catalyst surface.22–24 When cerium was added to Cu4/ZSM-5, a broad band centered at 300 nm appeared, which can be attributed to well-dispersed Ce species on the surface of catalyst (presumably in a tetra-coordinated environment), which is confirmed by the XRD results.25 Meanwhile, the intensity of 258 nm band was increased compared with that in UV-Vis spectra of Cu4/ZSM-5 catalyst, indicating the increase in the amount of isolated Cu2+ ions, which is consistent with the XPS results. On the contrary, it is difficult to quantify CuO crystallites on the catalyst surface because two bands centered at 300 and 750 nm overlap complexity.
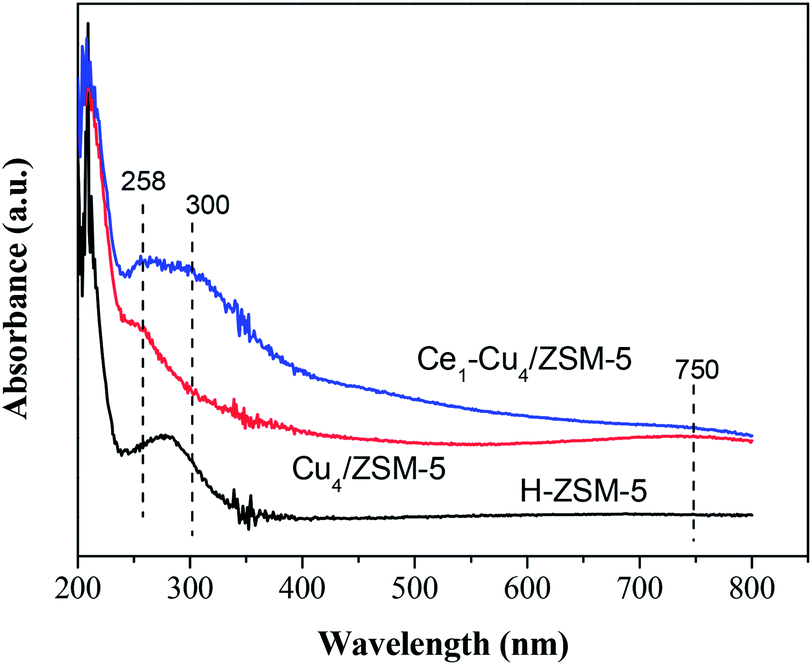 | ||
| Fig. 5 UV-Vis spectra of H-ZSM-5, Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts. | ||
3.3 Temperature-programmed desorption
Fig. 6 shows NH3-TPD profiles of the H–ZSM-5, Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts in the temperature range of 100–500 °C. Two desorption peaks at 200 and 410 °C can be observed for the H–ZSM-5 catalyst. The desorption peak at 200 °C is assigned to physisorbed NH3 or ammonium species, and the desorption peak at 410 °C is assigned to NH3 absorbed at the strong acid sites.26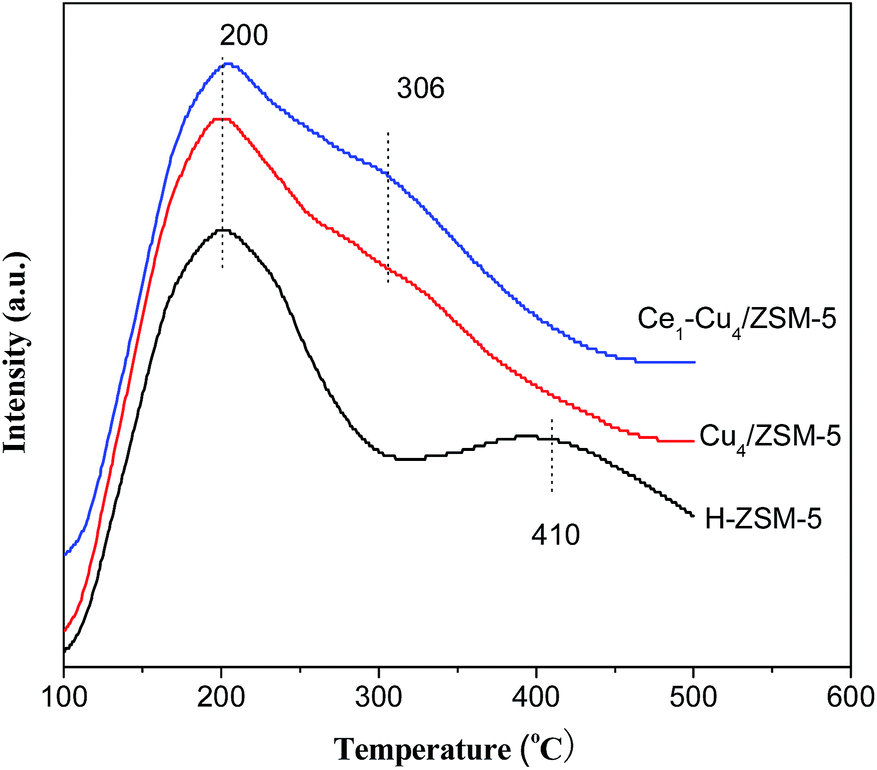 | ||
| Fig. 6 NH3-TPD profiles of H–ZSM-5, Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts. | ||
After adding copper and cerium, the NH3-TPD profiles have been obviously changed. The desorption peak at 410 °C radically decreased for the Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts. On the contrary, a shoulder peak at 306 °C presented for the Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts. The reasons for this phenomena were because part of Brønsted acid protons were substituted by metal ion and strong Lewis acid sites were produced originating from metal oxide nanoparticles when adding Cu and/or Ce into the H–ZSM-5 catalyst.27,28 Compared with the Cu4/ZSM-5 catalyst, the peak area at 306 °C is slightly larger for the Ce1–Cu4/ZSM-5 catalyst, even if the specific surface area was considered, because the BET surface area (268 m2 g−1) of the former is less than that (280 m2 g−1) of the latter, indicating that the Ce1–Cu4/ZSM-5 catalyst possessed the more strong acid sites than that on the Cu4/ZSM-5 catalyst.
Fig. 7 shows IR spectra of adsorbed pyridine on H–ZSM-5, Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts. The absorption bands at 1450 cm−1 and 1545 cm−1 are assigned to Lewis acid sites and Brønsted acid sites respectively, while the absorption band at 1490 cm−1 is assigned to both of them. The concentration of the Lewis acid sites (LA) and Brønsted acid sites (BA) were calculated from the intensity of the absorption bands at 1450 cm−1 and 1545 cm−1, and the results are shown in Table 2. It can be seen that the H–ZSM-5 catalyst mainly exhibited Brønsted acid sites. After adding copper, the amount of Brønsted acid sites markedly decreased due to the ion exchange during the preparation step. On the contrary, the amount of Lewis acid sites increased largely because of the presence of Cu2+ ions on the surface. Furthermore, compared with the H–ZSM-5 catalyst, the Cu4/ZSM-5 catalyst possessed the higher amount of total acid sites. When adding cerium into the Cu4/ZSM-5 catalyst, the amounts of both the Lewis acid sites and Brønsted acid sites were increased, leading to the higher amount of total acid sites, which is in accordance with the NH3-TPD results. It has been proved that Brønsted acid sites may not be required for activating ammonia, while Lewis acid sites plays an important role on the NH3-SCR process catalyzed by the zeolite catalyst.29 Therefore, the more Lewis acid sites over the Ce1–Cu4/ZSM-5 catalyst might make the higher NH3-SCR activity of Ce1–Cu4/ZSM-5 than the Cu4/ZSM-5 catalyst.
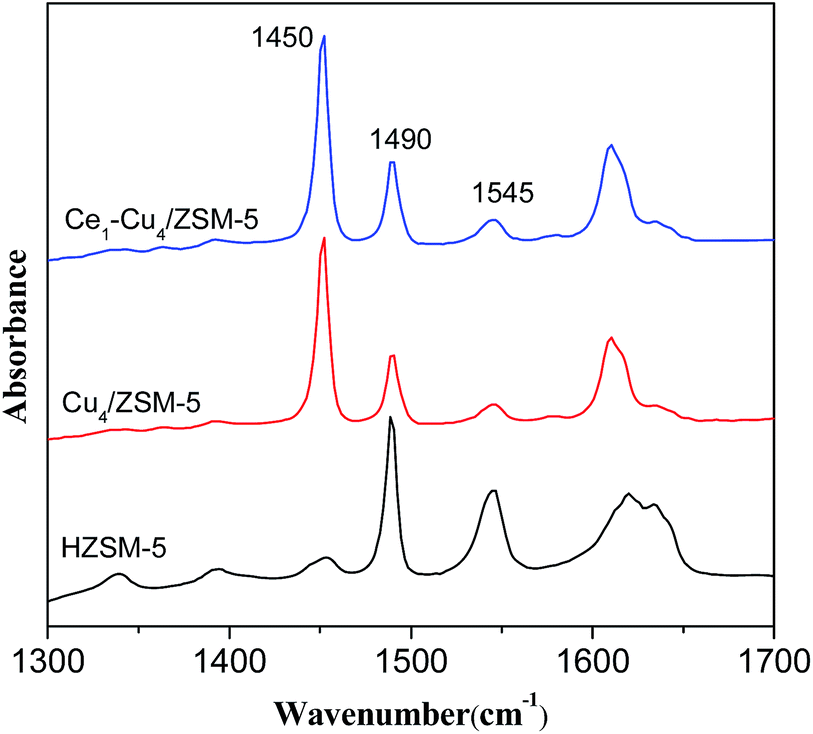 | ||
| Fig. 7 IR spectra of adsorbed pyridine on H–ZSM-5, Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts. | ||
The NO-TPD profiles of the catalysts are also shown in Fig. 8. Two obvious desorption peaks centered at 120 and 400 °C presented in the NO-TPD profile of the Cu4/ZSM-5 catalyst. The desorption peak at 120 °C can be attributed to physisorbed NOx, and the desorption peak at 400 °C is due to the decomposition of nitrite and nitrate species with higher thermal stability.30 After adding Ce in the Cu4/ZSM-5 catalyst, the desorption peak area at 400 °C increased remarkably, indicating that the addition of Ce can enhance the adsorption of nitrite and nitrate species on the surface of the Cu4/ZSM-5 catalyst. In addition, the presence of Ce in this catalyst also improves the adsorption capacity and ability for NO at low temperature. As a result, more nitrite and nitrate species on the Ce1–Cu4/ZSM-5 catalyst surface can participate the SCR reaction than that on the surface of the Cu4/ZSM-5 catalyst, and the Ce doped catalyst can adsorb NO at lower temperature, resulting in the enhancement of the catalytic activity and the extension of the operation temperature window of the Ce1–Cu4/ZSM-5 catalyst.
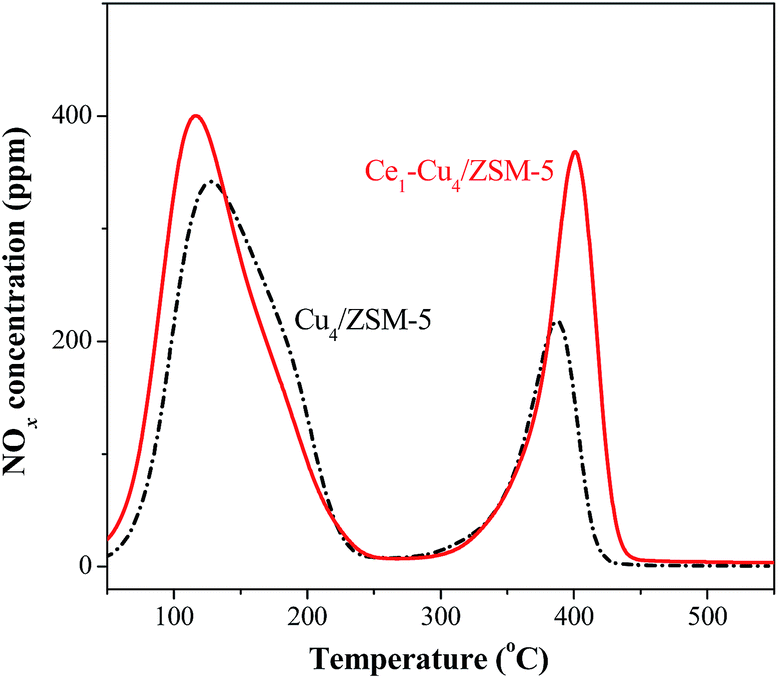 | ||
| Fig. 8 NO-TPD profiles of the Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts. | ||
3.4 H2-TPR
Fig. 9 shows H2-TPR profiles of H–ZSM-5, Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts. The H–ZSM-5 catalyst does not exhibit any reduction peaks. There are three reduction peaks at 213, 245 and 330 °C in the TPR profile of the Cu4/ZSM-5 catalyst. The reduction peak at 213 °C is attributed to the reduction of CuO nanoparticles dispersed on the ZSM-5 surface to Cu0, and the reduction peaks at 245 and 330 °C are assigned to the reduction of isolated Cu2+ to Cu0 through two steps.31 The reduction Cu2+ → Cu+ can occur at lower temperature, and the reduction Cu+ → Cu0 only can carry out at higher temperature. For the Ce1–Cu4/ZSM-5 catalyst, there are also three reduction peaks, the peak at 330 °C shifted to a higher temperature of 350 °C, and the reduction peak area at 213 °C was markedly increased comparing with the Cu4/ZSM-5 catalyst, due to the high surface concentrations of CuO nanoparticles (Table 1). The results above show that adding Ce in the Cu4/ZSM-5 catalyst can enhance its reducibility.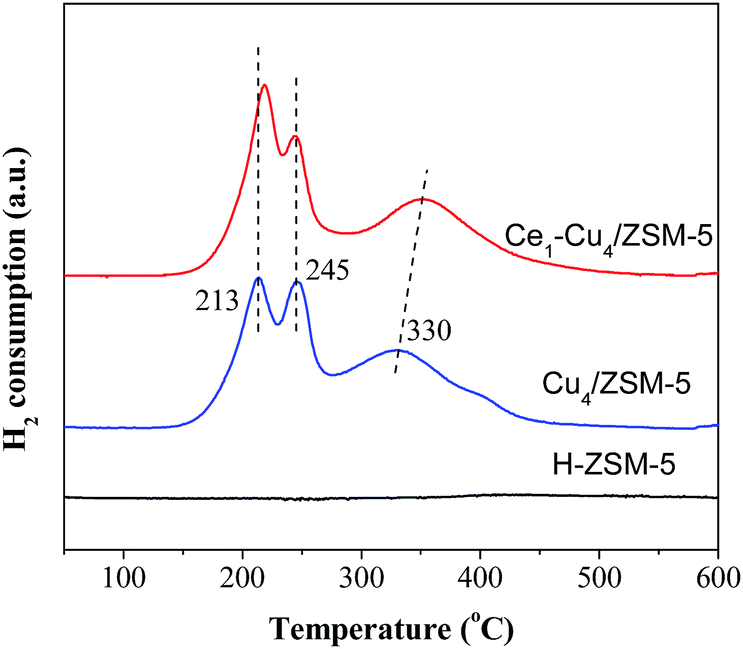 | ||
| Fig. 9 H2-TPR profiles of H–ZSM-5, Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts. | ||
3.5 NO oxidation
It is well known that the improvement of NO oxidation to NO2 over SCR catalysts can significantly promote its low temperature activity, due to the occurrence of the “fast SCR”, NO + NO2 + 2NH3 → 2N2 + 3H2O.32 Therefore, the effect of adding Ce on the catalytic activity of the Cu4/ZSM-5 catalyst for NO oxidation has been investigated. As shown in Fig. 10, for the NO oxidation at <400 °C, the activity of the Ce1–Cu4/ZSM-5 catalyst is slightly higher than that of the Cu4/ZSM-5 catalyst, due to the higher reducibility of the Ce1–Cu4/ZSM-5 catalyst. At the high temperature, there is no difference of the catalytic activity of both catalysts for NO oxidation because of the equilibrium conversion.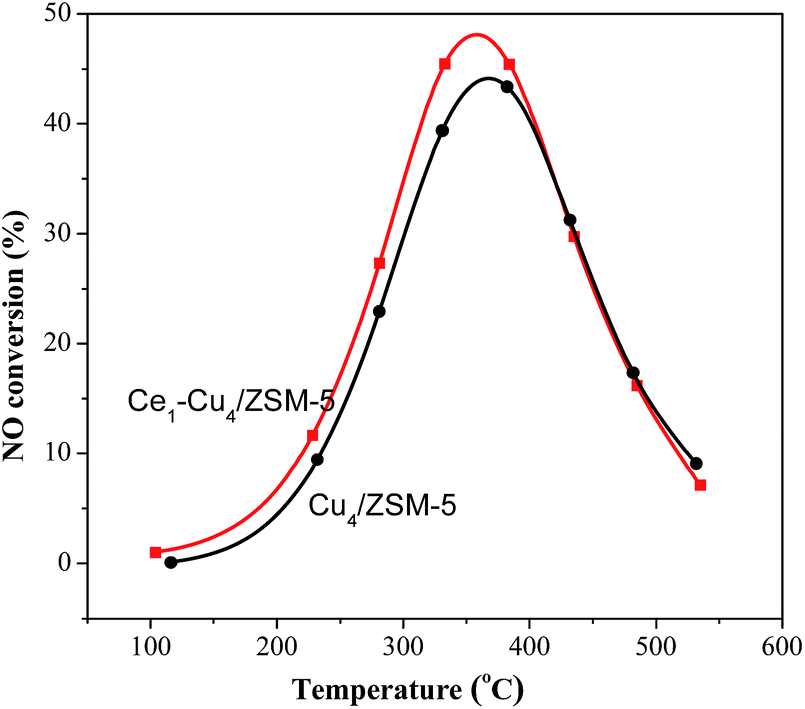 | ||
| Fig. 10 NO conversion in separate NO oxidation reaction over the Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts. (Reaction conditions: 500 ppm NO + 5 vol% O2/Ar balanced, total flow rate 300 mL min−1). | ||
3.6 In situ DRIFT spectroscopy
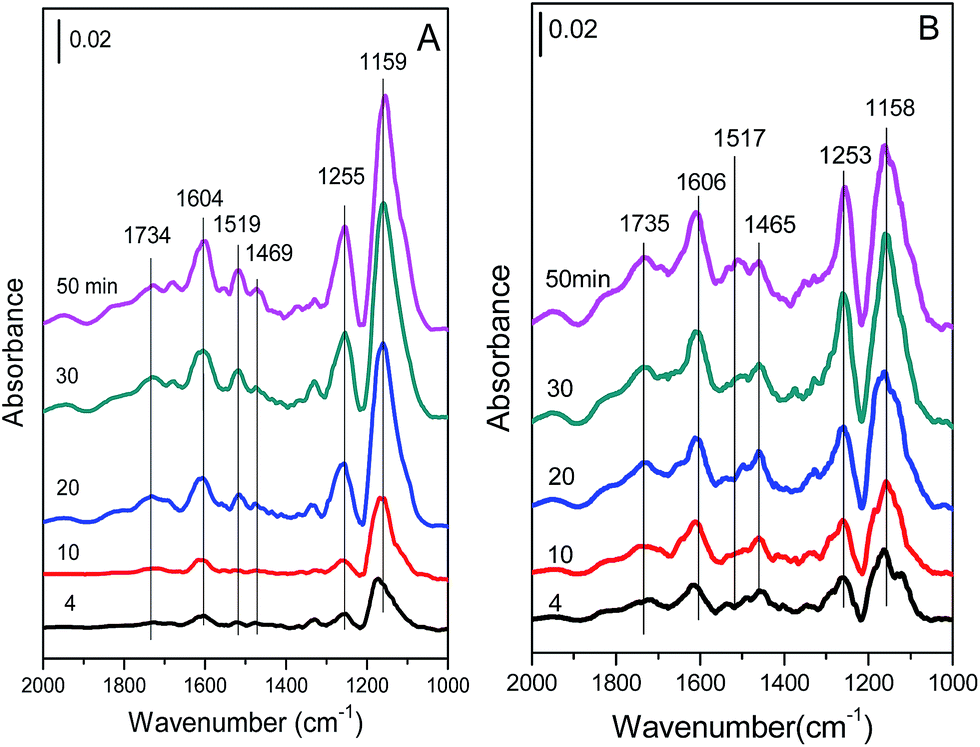 | ||
| Fig. 11 In situ DRIFT spectra of (A) Cu4/ZSM-5 and (B) Ce1–Cu4/ZSM-5 catalysts exposed to a flow of 500 ppm NH3/Ar (50 mL min−1) at 150 °C for different times. | ||
The bands at 1159 and 1255, 1604 cm−1 are assigned to coordinated NH3 adsorbed on Lewis acid sites, the bands at 1469 and 1734 cm−1 are assigned to NH4+ ions located on Brønsted acid sites.19,33–36 And the band at 1519 cm−1 might be attributed to amide (–NH2) species.19,37 With an increase in the exposure time, the intensities of all bands are obviously increased, indicating the increase in the amount of adsorbed NH3 species. After adding Ce in the Cu4/ZSM-5 catalyst, its in situ DRIFT spectra are similar to that of the Cu4/ZSM-5 catalyst. The coordinated NH3 adsorbed on Lewis acid sites (bands at 1158, 1253 and 1606 cm−1), and NH4+ ions located on Brønsted acid sites (bands at 1469 and 1734 cm−1) are also presented, and their intensities are increased obviously with the increase in the exposure time of NH3. As shown in Fig. 11, Lewis acid sites on the two catalysts are dominant to adsorb or activate NH3 compared with Brønsted acid sites, because most of Brønsted acid sites have been substituted by Ce or Cu ions.38
After the experiments of Fig. 11 were finished, the catalyst was purged with Ar (50 mL min−1) for 1 h, and then was exposed to 500 ppm NO + 5 vol% O2/Ar (50 mL min−1). In situ DRIFT spectra of the catalyst were taken for different times and are presented in Fig. 12.
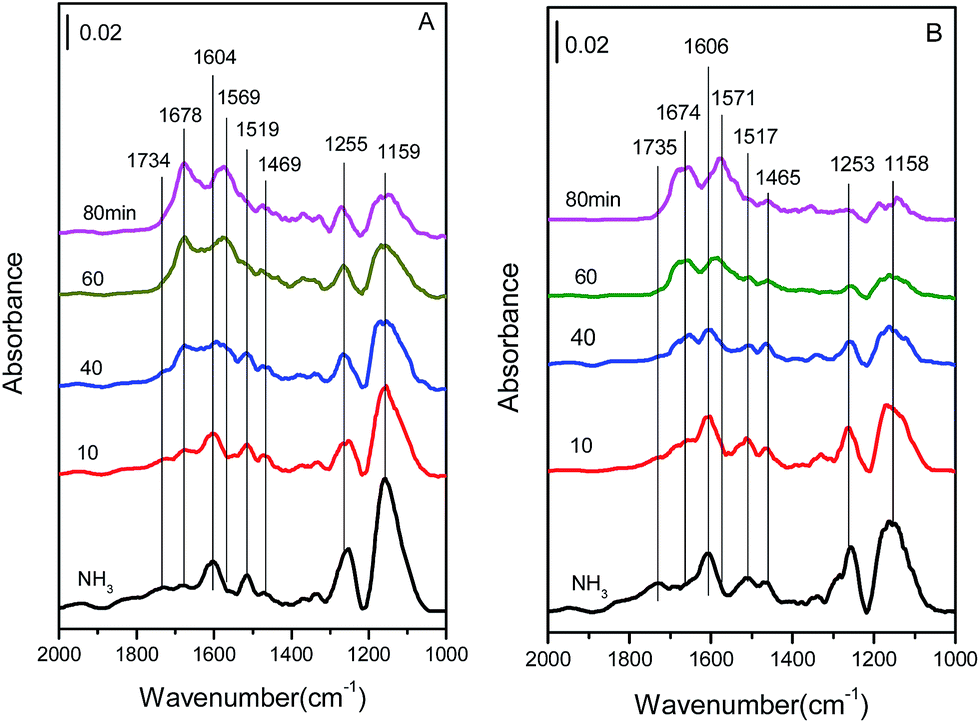 | ||
| Fig. 12 In situ DRIFT spectra of (A) Cu4/ZSM-5 and (B) Ce1–Cu4/ZSM-5 catalysts in the mixed gas of 500 ppm NO + 5 vol% O2/Ar at different times, after adsorption of 500 ppm NH3/Ar at 150 °C and blowing by Ar for 1 h. | ||
As shown in Fig. 12A, when NO + O2 is exposed to NH3-pretreated Cu4/ZSM-5 catalyst, the intensities of the bands at 1159, 1255, 1604, 1519 and 1734 cm−1 decreased gradually with the increase in the exposure time of NO + O2, which indicates that both NH3 species adsorbed on Lewis acid sites and NH4+ ions located on Brønsted acid sites are involved in the NH3-SCR reaction. At the same time, many new bands at 1678 and 1569 cm−1 ascribed to nitrates species appeared and increased with the increase of the exposure time in NO + O2. After adding Ce, the bands assigned to adsorbed NH3 species decreased more quickly on the Ce1–Cu4/ZSM-5 catalyst with the increase in the exposure time of NO + O2 (Fig. 12B), indicating the higher SCR activity compared with the Cu4/ZSM-5 catalyst.
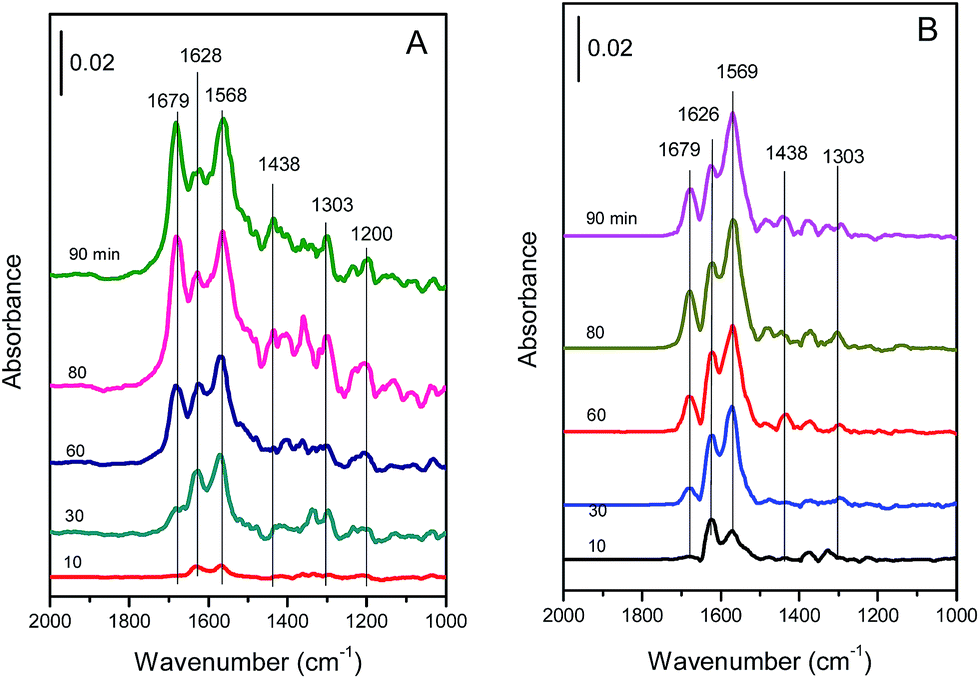 | ||
| Fig. 13 In situ DRIFT spectra of the (A) Cu4/ZSM-5 and (B) Ce1–Cu4/ZSM-5 catalysts exposed to a flow of 500 ppm NO + 5 vol% O2/Ar (50 mL min−1) at 150 °C for different times. | ||
As shown in Fig. 13A, there are three strong bands at 1679, 1628 and 1568 cm−1 in the DRIFT spectra of the Cu4/ZSM-5 catalyst. The bands at 1628 and 1568 cm−1 are assigned to bridged and bidentate nitrates respectively,39–42 and the band at 1679 cm−1 is attributed to ionic nitrite species.43,44 These bands were increased with the exposure time. Besides, there are still three bands with low intensity at 1438, 1303 and 1200 cm−1 in the DRIFT spectra of the Cu4/ZSM-5 catalyst. These bands are assigned to nitrite species.42,44,45
Compared with the in situ DRIFT spectra of the Cu4/ZSM-5 catalyst, the strong absorption bands at 1679, 1626 and 1569 cm−1 are also shown in the in situ DRIFT spectra of the Ce1–Cu4/ZSM-5 catalyst (Fig. 13B), but the intensity of the absorption band at 1679 cm−1 corresponding to ionic nitrite species is much lower than those at 1626 and 1569 cm−1. This is because that the higher oxidation of the Ce1–Cu4/ZSM-5 catalyst (as shown in H2-TPR results) can easily oxidize ionic nitrite species to nitrates, resulting in the reduction in the ionic nitrite species on the Ce1–Cu4/ZSM-5 catalyst surface. At the same time, the intensities of the bands at 1438 and 1303 cm−1 are much low, and the band at 1200 cm−1 assigned to bridged nitrite has even disappeared. Since nitrates are the active species for the SCR reaction, the highly catalytic activity of the Ce1–Cu4/ZSM-5 catalyst for the oxidation of nitrite to nitrate is favorable for the better SCR activity.
To study the reaction between NH3 and adsorbed NOx species, after the experiments of Fig. 13 were finished, the catalyst was purged with Ar (50 mL min−1) for 1 h, and then was exposed to 500 ppm NH3/Ar (50 mL min−1). In situ DRIFT spectra of the catalyst were taken for different times and their results are presented in Fig. 14.
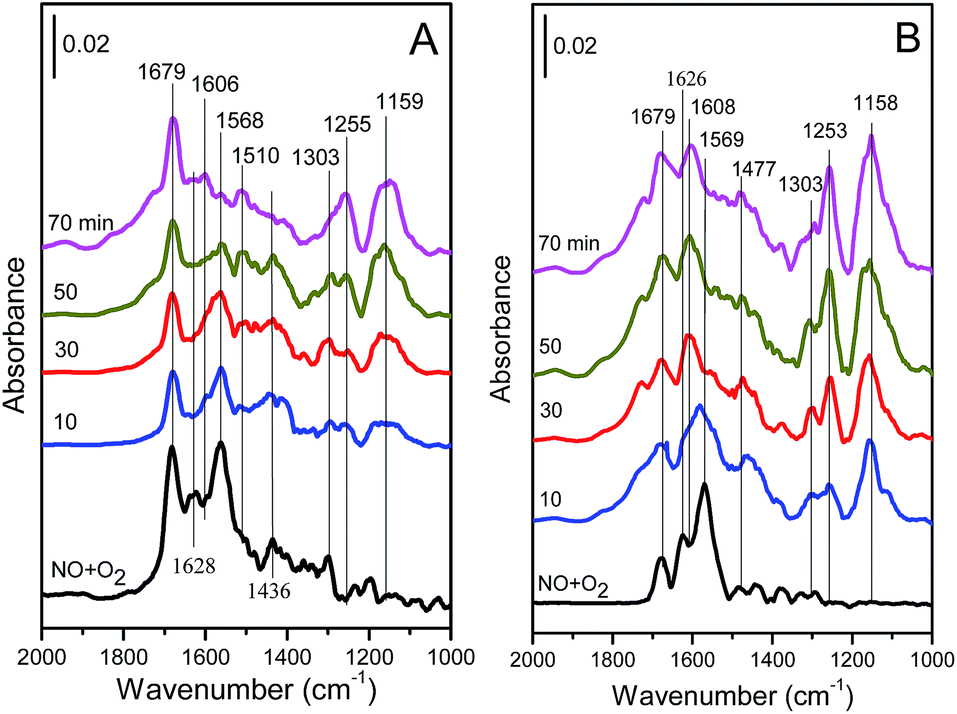 | ||
| Fig. 14 In situ DRIFT spectra over (A) Cu4/ZSM-5 and (B) Ce1–Cu4/ZSM-5 catalysts in 500 ppm NH3/Ar for different times after adsorption of 500 ppm NO + 5 vol% O2/Ar at 150 °C and blowing by Ar for 1 h. | ||
As shown in Fig. 14A, the intensities of the bands at 1568 cm−1 (assigned to bidentate nitrate) and 1628 cm−1 (assigned to bridged nitrate) decreased quickly with the increase of the exposure time in 500 ppm NH3, indicating that bidentate and bridged nitrates are the reactive species with a high activity for the SCR reaction. On the contrast, the intensity of band at 1679 cm−1 (assigned to ionic nitrite species) was hardly changed even after NH3 was passed over the catalyst for 80 min, showing that this species is inactive in SCR process. Simultaneously, the bands at 1159, 1255 and 1606 cm−1 originating from NH3 adsorption species appear, and their intensities increase with the increase of the exposure time in NH3.
Like the DRIFT results of the Cu4/ZSM-5 catalyst, the intensities of the bands at 1569 cm−1 and 1626 cm−1 over the Ce1–Cu4/ZSM-5 catalyst decreased with the increase in the exposure time of NH3, and these two bands disappeared after 30 min, which is less than that for the disappearance of these two bands over the Cu4/ZSM-5 catalyst (60 min), indicating a high SCR activity of the Ce1–Cu4/ZSM-5 catalyst. On the contrary, the variation of the band at 1679 cm−1 was unobvious with the time. At the same time, the bands at 1158, 1253, 1477 and 1608 cm−1 originating from NH3 adsorption species appeared, and their intensities increased with the increase of the exposure time in NH3. Compared with the DRIFT results of the Cu4/ZSM-5 catalyst, the intensities of the bands assigned to NH3 adsorption species over the Ce1–Cu4/ZSM-5 catalyst are higher.
For the Ce1–Cu4/ZSM-5 catalyst, the bands assigned to adsorbed NH3 species on the Ce1–Cu4/ZSM-5 catalyst was decreased more quickly with the increase in the exposure time of NO + O2 (Fig. 12B), indicating its higher SCR activity than the Cu4/ZSM-5 catalyst. As shown in Fig. 13, the NO species can adsorb on the surface of the Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts in the forms of bridged and bidentate nitrates, ionic nitrite and nitrate, and bridged nitrite. The nitrite species tend to be oxidized to nitrate species over the Ce1–Cu4/ZSM-5 catalyst, because of its high catalytic activity for the oxidation reaction. When the catalysts pretreated with the mixed gas of NO and O2 were exposed to NH3, bidentate nitrate and bridged nitrate were consumed very quickly, while ionic nitrite species was hardly changed. These results indicate that bidentate nitrate and bridged nitrate over the Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts are the reactive species with a high SCR reactivity, and the SCR reaction is followed Langmuir–Hinshelwood mechanism over the Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts.
As above mentioned, when the catalysts pretreated with the mixed gas of NO and O2 were exposed to NH3, bidentate nitrate and bridged nitrate consumed very quickly and disappeared after 30 or 60 min (Fig. 14). However, when the catalysts pretreated with NH3 were exposed to the mixed gas of NO and O2, the bands corresponding to NH3 adsorption in in situ DRIFT spectra (Fig. 12) decreased gradually, and a part of NH3 species adsorbed on the catalysts surface remained still after 80 min. Therefore, when the catalysts pretreated with NH3 were exposed to the mixed gas of NO and O2, NO maybe adsorbed on the surface of catalysts pretreated with NH3 firstly and then reacted with pre-adsorbed NH3, resulting in the low reaction rate. Therefore, it can't be confirmed whether the SCR reaction over the Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts is followed Eley–Rideal mechanism.
3.7 Effect of SO2 and H2O on the SCR reaction
Fig. 15 shows the effect of SO2 and H2O on the catalytic activities of the Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts for the SCR reaction at 200 °C. Before adding H2O or SO2, NOx conversion was 100% over the two catalysts and their activities was unchanged after 20 h of the reaction at 200 °C. When 2% H2O was introduced into the feed gas, NOx conversion over the Cu4/ZSM-5 catalysts immediately decreased to 90.2% after 8 h of the reaction. After 2% H2O in the feed gas was removed and lasting for 2 h, the NOx conversion was returned to ∼100%. After adding Ce in the Cu4/ZSM-5 catalyst, its performance for H2O resistance was improved, for instance, the NOx conversion could hold ∼94% in the presence of 2% water. And its NOx conversion was returned to ∼100% after removing 2% H2O in the feed gas and lasting for 1.5 h. A similar trend can also be observed at high reaction temperature (450 °C) over the Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts (Fig. S2†).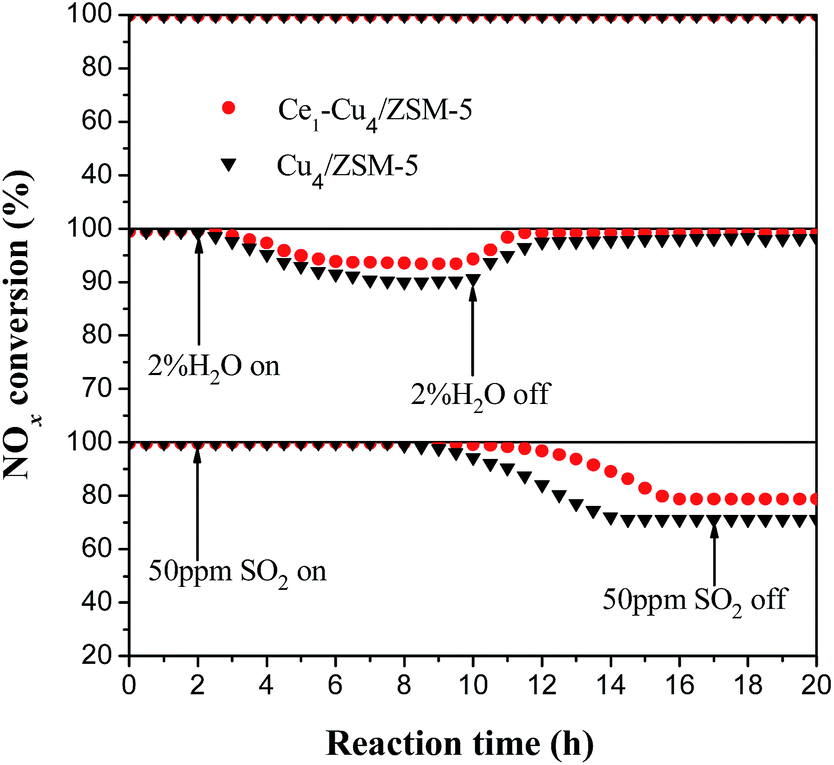 | ||
| Fig. 15 Effect of SO2 and H2O on the catalytic activities of Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts for SCR reaction at 200 °C. (Reaction conditions: 0.2 g catalyst, 500 ppm NO, 500 ppm NH3, 5% O2, Ar to balance, GHSV = 55000 h−1). | ||
When 50 ppm SO2 was introduced into the feed gas, 100% NOx conversion over the Cu4/ZSM-5 catalysts was kept for 8 h and then decreased to 70.4% after the reaction of 6 h, but the deactivation of catalyst poisoned by SO2 could not be recovered. However, it is obvious that the catalytic activity of the Ce1–Cu4/ZSM-5 catalyst was less affected by SO2 than the Cu4/ZSM-5 catalyst, that is to say, the Ce doping can improve the SO2 resistance of the Cu4/ZSM-5 catalyst. A similar trend can also be observed at high reaction temperature over the Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts (Fig. S2†). It is well known that the ammonium sulfate and copper sulfate deposited on the catalyst surface can cover available active sites and block zeolite channels,46 and they could not be decomposed and removed at 200 °C on the catalyst surface,47 resulting in a deactivation of catalyst. The presence of Ce in the Cu4/ZSM-5 catalyst can make SO2 firstly deposit on the Ce site to form a stable Ce sulfate, which can inhibit the formation of ammonium sulfate and copper sulfate on the catalyst surface in the SCR process, improving the excellent sulfur tolerance of the Ce1–Cu4/ZSM-5 catalyst.
4. Conclusions
In summary, the addition of Ce can markedly improve the catalytic activity of the Cu/ZSM-5 catalyst for NH3-SCR reaction and enhance its SO2 and water resistance. Adding Ce in the Cu4/ZSM-5 catalyst promoted the enrichment of copper species on the catalyst surface and increased the surface concentrations of both isolated Cu2+ ions and CuO crystallites, resulting in the improvement of the redox properties and the NO adsorption ability of the Cu4/ZSM-5 catalyst. The Ce amount would affect the catalytic performance of Cex–Cu/ZSM-5, especially the NH3-SCR activity at high temperature. When the Ce1–Cu4/ZSM-5 catalyst (Ce/Cu = 1/4, wt) was used, the temperature window for more than 90% NOx conversion was extended to 185–470 °C, which broadened the operation window about 45 °C, because more nitrite and nitrate species on the Ce1–Cu4/ZSM-5 catalyst surface can participate the SCR reaction than that on the surface of the Cu4/ZSM-5 catalyst, and the Ce-doping sample can adsorb NO at lower temperature. In situ DRIFTS results indicated that the Langmuir–Hinshelwood mechanism for the NH3-SCR reaction over the Cu4/ZSM-5 and Ce1–Cu4/ZSM-5 catalysts was confirmed, but Eley–Rideal mechanism can't be sure yet.Acknowledgements
This work was supported financially by the National Natural Science Foundation of China (21577034, 21333003), the Commission of Science and Technology of Shanghai Municipality (15DZ1205305) and the Fundamental Research Funds for the Central Universities (WJ1514020).Notes and references
- A. Brückner, F. Hipler, G. Auer, E. Löffler and W. Grünert, J. Catal., 2012, 286, 237 CrossRef.
- Z. M. Liu, J. H. Li and S. I. Woo, Energy Environ. Sci., 2012, 5, 8799 CAS.
- W. P. Shan, F. D. Liu, H. He, X. Y. Shi and C. B. Zhang, Catal. Today, 2012, 184, 160 CrossRef CAS.
- M. J. Li, Y. Yeom, E. Weitz and W. M. H. Sachtler, J. Catal., 2005, 235, 201 CrossRef CAS.
- X. F. Yang, Z. L. Wu, M. Moses-Debusk, D. R. Mullins, S. M. Mahurin, R. A. Geiger, M. Kidder and C. K. Narula, J. Phys. Chem. C, 2012, 116, 23322 CAS.
- H. Sjövall, L. Olsson, E. Fridell and R. J. Blint, Appl. Catal., B, 2006, 64, 180 CrossRef.
- G. Carja, Y. Kameshima, K. Okada and C. D. Madhusoodana, Appl. Catal., B, 2007, 73, 60 CrossRef CAS.
- J. L. de Lucas, F. Valverde, A. Dorado and I. Romero, J. Mol. Catal. A: Chem., 2005, 225, 47 CrossRef.
- J. H. Park, H. J. Park, J. H. Baik, I. S. Nam, C. H. Shin, J. H. Lee, B. K. Cho and S. H. Oh, J. Catal., 2006, 240, 47 CrossRef CAS.
- X. F. Tang, Y. G. Li, X. M. Huang, Y. D. Xu, H. Q. Zhu, J. G. Wang and W. J. Shen, Appl. Catal., B, 2006, 62, 265 CrossRef CAS.
- G. Carja, Y. Kameshima, K. Okada and C. D. Madhusoodana, Appl. Catal., B, 2007, 73, 60 CrossRef CAS.
- Z. B. Wu, R. B. Jin, Y. Liu and H. Q. Wang, Catal. Commun., 2008, 9, 2217 CrossRef CAS.
- S. M. Lee, K. H. Park and S. C. Hong, Chem. Eng. J., 2012, 195–196, 323 CrossRef CAS.
- K. Liu, F. D. Liu, L. J. Xie, W. P. Shan and H. He, Catal. Sci. Technol., 2015, 5, 2290 CAS.
- Y. P. Zhang and M. Flytzani-Stephanopoulos, J. Catal., 1996, 164, 131 CrossRef CAS.
- J. Y. Yan, W. M. H. Sachtler and H. H. Kung, Catal. Today, 1997, 33, 279 CrossRef CAS.
- L. Pang, C. Fan, L. N. Shao and K. P. Song, Chem. Eng. J., 2014, 253, 394 CrossRef CAS.
- B. J. Dou, G. Lv, C. Wang, Q. Hao and K. Hui, Chem. Eng. J., 2015, 270, 549 CrossRef CAS.
- F. Bin, Appl. Catal., B, 2014, 150–151, 532 CrossRef CAS.
- B. Pereda-Ayo, U. D. L. Torre, M. J. Illán-Gómez, A. Bueno-López and J. R. González-Velasco, Appl. Catal., B, 2014, 147, 420 CrossRef CAS.
- L. Wang, J. R. Gaudet, W. Li and D. Weng, J. Catal., 2013, 306, 68 CrossRef CAS.
- M. H. Groothaet, J. A. van Bokhoven, A. A. Battiston, B. M. Weckhuysen and R. A. Schoonheydt, J. Am. Chem. Soc., 2003, 125, 7629 CrossRef PubMed.
- T. Zhang, J. Liu, D. Wang, Z. Zhao, Y. Wei, K. Cheng, G. Jiang and A. Duan, Appl. Catal., B, 2014, 148–149, 520 CrossRef CAS.
- S. Yashnik and Z. Ismagilov, Appl. Catal., B, 2015, 170–171, 241 CrossRef CAS.
- Y. Fu, W. Zhan, Y. Guo, Y. Wang, X. Liu, Y. Guo, Y. Wang and G. Lu, Microporous Mesoporous Mater., 2015, 214, 101 CrossRef CAS.
- T. Zhang, J. Liu, D. X. Wang, Z. Zhao, Y. C. Wei, K. Cheng, G. Y. Jiang and A. J. Duan, Appl. Catal., B, 2014, 148–149, 520 CrossRef CAS.
- G. H. Kuehl and H. K. C. Timken, Microporous Mesoporous Mater., 2000, 35–36, 521 CrossRef CAS.
- L. J. Lobree, I. C. Hwang and J. A. Reimer, J. Catal., 1999, 186, 242–253 CrossRef CAS.
- X. Liang, J. X. Li, Q. C. Lin and K. Q. Sun, Catal. Commun., 2007, 8, 1901 CrossRef CAS.
- X. D. Wu, F. Lin, H. B. Xu and D. Weng, Appl. Catal., B, 2010, 96, 101 CrossRef CAS.
- A. de Lucas, J. L. Valverde, F. Dorado, A. Romero and I. Asencio, J. Mol. Catal. A: Chem., 2005, 225, 47 CrossRef CAS.
- J. Chen, M. Shen, X. Wang, G. Qi, J. Wang and W. Li, Appl. Catal., B, 2013, 251, 134 Search PubMed.
- L. Wang, W. Li, S. J. Schmieg and D. Weng, J. Catal., 2015, 324, 98 CrossRef CAS.
- F. Gao, J. H. Kwak, J. Szanyi and C. H. F. Peden, Top. Catal., 2013, 56, 1441 CrossRef CAS.
- H. Y. Zhu, J. H. Kwak, C. H. F. Peden and J. Szanyi, Catal. Today, 2013, 205, 16 CrossRef CAS.
- L. Wang, W. Li, G. S. Qi and D. Weng, J. Catal., 2012, 289, 21 CrossRef CAS.
- W. S. Kijlstra, D. S. Brands, H. I. Smit, E. K Poels and A. Bliek, J. Catal., 1997, 171, 219 CrossRef CAS.
- S. Brandenberger, O. Kröcher, A. Wokaun, A. Tissler and R. Althoff, J. Catal., 2009, 268, 297 CrossRef CAS.
- W. Shan, F. Liu, H. He, X. Shi and C. Zhang, Appl. Catal., B, 2012, 115–116, 100 CrossRef CAS.
- S. Liu, X. D. Wu, D. Weng and R. Rani, Ind. Eng. Chem. Res., 2012, 51, 2271 CrossRef.
- R. D. Zhang, W. Yang, N. Luo, P. X. Li, Z. G. Lei and B. H. Chen, Appl. Catal., B, 2014, 146, 94 CrossRef CAS.
- J. Szanyi, J. H. Kwak, H. Y. Zhu and C. H. F. Peden, Phys. Chem. Chem. Phys., 2013, 15, 2368 RSC.
- K. Hadjiivanov, A. Penkova, M. Daturi, J. Saussey and J. C. Lavalley, Chem. Phys. Lett., 2003, 377, 642 CrossRef CAS.
- K. I. Hadjiivanova, Catal. Rev.: Sci. Eng., 2000, 42, 71 Search PubMed.
- I. Atribak, B. Azambre, A. Bueno López and A. García-García, Appl. Catal., B, 2009, 92, 126 CrossRef CAS.
- Z. X. Ma, H. S. Yang, F. Liu and X. B. Zhang, Appl. Catal., A, 2013, 467, 450 CrossRef CAS.
- J. Yu, F. Guo, Y. L. Wang, J. H. Zhu, Y. Y. Liu and F. B. Su, Appl. Catal., B, 2010, 95, 160 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra12505g |
| This journal is © The Royal Society of Chemistry 2015 |