
Simple synthesis 11-substituted norcryptotackieine derivatives†
Abstract
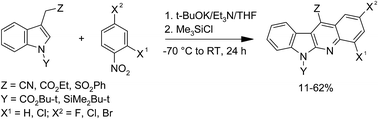
11-Substituted (CN, CO2Et, SO2Ph) indolo[2,3-b]quinolines were obtained in reactions of N-protected (SiMe2t-Bu, CO2t-Bu) indol-3-yl-acetonitrile, -acetate, and -methylsulfone with nitrobenzene derivatives in presence of base and trialkylchlorosilanes.
 Please wait while we load your content...
Please wait while we load your content...

