
Identification of the over alkylation sites of a protein by IAM in MALDI-TOF/TOF tandem mass spectrometry†
Mengzhe Guo‡
abc,
Guofeng Weng‡a,
Dengyang Yinbc,
Xunxiu Hubc,
Jie Hanbc,
Yan Dubc,
Yaqin Liua,
Daoquan Tang*bc and
Yuanjiang Pan*a
aDepartment of Chemistry, Zhejiang University, Zheda road 38#, Hangzhou 310027, China. E-mail: panyuanjiang@zju.edu.cn; Fax: +86-571-87951629; Tel: +86-571-87951264
bKey Laboratory of New Drug Research and Clinical Pharmacy, Xuzhou Medical College, Xuzhou, Jiangsu 221004, China
cDepartment of Pharmaceutical Analysis, Xuzhou Medical College, Xuzhou, Jiangsu 221004, China
First published on 18th November 2015
Abstract
Rationale: overalkylation often appears during the proteolytic digestion process when using iodoacetamide (IAM) to protect the produced side chain thiol of Cys from disulfide bonds. However, side reactions between IAM and other functional groups of amino acids have not been investigated completely. Here, matrix-assisted laser desorption ionization equipped with time of fight analyzer tandem mass spectrometry (MALDI-TOF/TOF MS) has been used to identify overalkylation sites and compare the reactivity of different amino acid residues accurately and rapidly. Methods: in this work, H13A protein, an angiogenin mutant with a mutation from His to Ala at the thirteenth site was analyzed by the bottom-up method and the overalkylation sites were determined by MALDI tandem mass spectrometry. Results: MS results show two or more acetyl groups can be attached to the tryptic peptides. MS/MS results further show the attached acetyl is produced by a reaction between IAM and the side chain of Asp, Lys or His. This result indicates side reactions of IAM with other amino acids and the tendency seems to be Cys > His > Asp > Lys. Conclusions: besides the side chain thiol of Cys, the excess IAM could also react with the side chain of other amino acids such as Asp, Lys and His, etc. The clarified mechanism of this phenomenon can help to avoid disturbances of the interference peaks produced by side reactions and also contribute to the improvement of the method by accurately choosing tryptic peptides.
Introduction
Mass spectrometry has been developed as a fast and accurate technique in the determination and characterization of both unknown and complicated compounds.1,2 Particularly since the 21st century, mass spectrometry has been widely used in the identification of biological samples such as peptides and proteins with soft ionization techniques such as electrospray ionization (ESI) or matrix-assisted laser desorption ionization (MALDI).3–5 In protein analysis, there are two main approaches for the determination of the primary structure (amino acid sequence) by MS: “bottom-up method” and “top-down method”.6–8 The similarity of both approaches is turning the proteins into series types of fragment ions in mass spectrum and obtaining the sequence data by the analysis of these ions. Additionally compared to the top-down method, bottom-up method has been more widely applied in the protein analysis because of the minimal equipment requirements. In bottom-up method, analyses are undertaken at the peptide level after protein digestion, which is usually performed with trypsin.9 Tryptic peptides have the characteristic that they contain either arginine or lysine at the C-terminal. They also can be analyzed in the forms of either complex peptide mixture or isolated peptide by using different complementary mass spectrometry techniques such as MALDI-TOF/TOF and LC-ESI-MS/MS for the sequence information.10,11 By the comparison of cleavages of known archived proteins from a sequence database or annotated peptide spectral in a peptide spectral library, the proteolytic peptides can be indentified and final induced the protein identification.12 Besides, in most proteomic studies the proteolytic peptides also can be sequenced by collision-induced dissociation (CID) tandem mass spectrometry. The MS/MS results are usually analyzed by the database searches.13–15 Generally speaking, bottom-up method has been widely used in the research of peptides and proteins by MALDI mass spectrometry.16–18 For example, Rodthongkum et al. analyzed the enriched low abundance peptide biomarkers of human by MALDI-MS.19It is noted that before the protein digestion in bottom-up method, several preparations must be carried out to make the digestion completely. The disulfide bonds of the protein must be opened (usually by dithiothreitol, DTT) and the thiols from disulfide bond dissociation is usually protected by iodoacetamide (IAM).20,21 IAM can easily react with the thiol at room temperature. However, this alkylation often leads to unwanted side reactions.22 IAM also reacts with amines or side chain group of other amino acids, which causes trouble in the analysis of protein.23–25 Previous literatures have proved that IAM can react with amino acids such as Asp, Glu, His, Tyr, etc.26,27 And the difficulty degree of the reaction between IAM and different amino acids has also been proposed. These works would help to reduce the disturbance of the overalkylation when protein was digested with IAM.
In this work, we have used H13A protein as the target to investigate the influence of the overalkylation to the protein analysis. The H13A protein is an angiogenin mutant with the mutation of the thirteenth amino site from His to Ala in. It is a single chain protein with 123 amino acids (about MW 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400) and containing three disulfide bonds. The role of His-13 in angiogenin has been demonstrated closely relationship with the function of angiogenin as rebonucleolytic.28–31 Replacement of this residue by Ala decreases the function of angiogenin drastically. After the protein preparation, the tryptic peptides of H13A protein have been analyzed directly by MALDI-TOF/TOF MS.32 The MS result has showed there were some interference peaks besides the tryptic peptide peaks. These interference peaks have certain regularity and may be produced by two or more IAM linked to the peptide. MS/MS technique has been used to identify the attached sites of IAM and further confirm the reaction tendency of different amino acids. This work can help to complete the mechanism of overalkylation so as to reduce its effect in protein analysis.
400) and containing three disulfide bonds. The role of His-13 in angiogenin has been demonstrated closely relationship with the function of angiogenin as rebonucleolytic.28–31 Replacement of this residue by Ala decreases the function of angiogenin drastically. After the protein preparation, the tryptic peptides of H13A protein have been analyzed directly by MALDI-TOF/TOF MS.32 The MS result has showed there were some interference peaks besides the tryptic peptide peaks. These interference peaks have certain regularity and may be produced by two or more IAM linked to the peptide. MS/MS technique has been used to identify the attached sites of IAM and further confirm the reaction tendency of different amino acids. This work can help to complete the mechanism of overalkylation so as to reduce its effect in protein analysis.
Experimental
Materials
The dithiothreitol (DTT), iodoacetamide were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). α-Cyano-4-hydroxycinnamic acid (CHCA), 2,5-dihydroxy-benzoic acid (DHB) were obtained from Bruker Franzen Analytik GmbH (Germany). H13A protein, the angiogenin which has the mutation (H-A) in the 13th amino acid respectively, was provided by Pro. Xu Zhengping, School of Medicine, Zhejiang University.Preparations and digestion of protein
20 μg protein was added in 50 mM tris buffer with 50 μL DTT (10 mM). Then the mixed solution was heated in water bath at 37 °C. The reaction lasted 3–4 h. After this reaction, 50 μL iodoacetamide (10, 20, 40, 50, 80 mM respectively) was added in the protein solution to continue reaction for half which was kept in dark. All these preparations were purposed to break the disulfide bonds of the protein and protect the thiols which produced by the dissociation of disulfide bonds.After preparations 0.2 μg trypsin (1:100 with the protein) was added into the mixed solution and heated in water bath at 37 °C for 12 h to achieve the digestion of the protein.
Mass spectrometry analysis
MS experiments were performed by Bruker ultraflextreme MALDI-TOF/TOF mass spectrometer (Bruker Daltonik, Germany) equipped with a nitrogen UV laser (337 nm 2000 Hz). Matrix and target mixed solution (v/v: 1/1) was dropped onto the MALDI plate and analyzed in positive ion reflection mode with the mass range of m/z 50–4500 Da. Ion source voltage was 25 kV and linear detector voltage was 2.9 kV. Each point was collected using 1000 laser shots and results were analyzed by Bruker flexAnalysis software.Result and discussion
Full scan mass spectrometry analysis of the H13A protein after digestion
As shown in Fig. 1, the tryptic peptides of the H13A protein which produced by trypsin digestion were mostly distributed in the range between m/z 500 to m/z 2500. The relative standard deviations of each tryptic peptide and its derivatives have been listed in Table S1.† Taking into consideration that this protein is about 14000 MW and contains sixteen trypsin cleavage sites, mass spectrometry results indicated that the protein was digested almost completely. Additionally comparing these tryptic peptides from H13A with angiogenin whose sequence is known, it can be indicated that tryptic peptide (TP) 1 of H13A protein with m/z 1896.0 has 66 molecular weights less than the tryptic peptide “YTHFLTQHYDAKPQGR” of angiogenin. So this tryptic peptide of H13A protein probably referred to the mutation and the MS/MS result of this peptide will verify this assumption and further identify the mutation sites. Conspicuously there are three tryptic peptides, m/z 1953.0, m/z 2010.1, m/z 2067.1, which are 57 MW, 2 × 57 MW or 3 × 57 MW more than the mutation peptide respectively. These peptides can be divided into one group which was named the group 1.
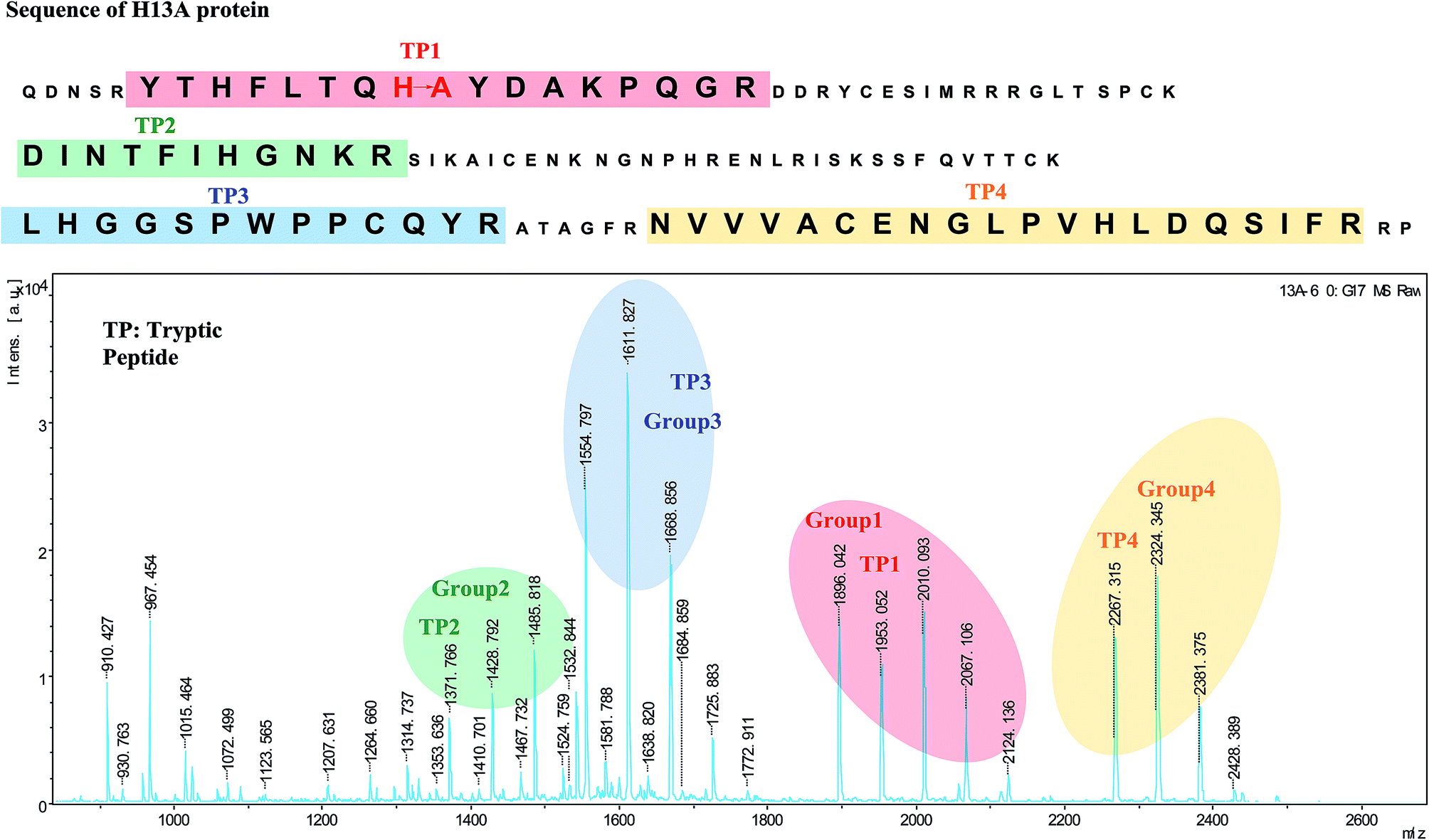 | ||
| Fig. 1 The tryptic peptides of the H13A protein in MALDI-TOF/TOF. | ||
Different concentrations of IAM have been reacted with the tryptic peptides to prove the overalkylation results mentioned above. Taking TP1 peptide as an example (shown in Fig. 2), it is obvious that the overalkylation degree of TP1 peptide has extended as the increase of the concentration of IAM. More specifically when the concentration of IAM increased from 10 mM to 20 mM, the peak intensity of TP1 + 57 enlarged and the peak of TP1 + 57 × 2 appeared. Additionally the peak of TP1 + 57 × 3 has been discovered when the concentration of IAM increased to 40 mM. Finally when the concentration increased to 80 mM the peak intensity of TP1 was even higher than that of TP1.
 | ||
| Fig. 2 The reactions between IAM with different concentrations and the sulfydryl of TP1 after reduction by 50 mM DTT, the reaction time is 0.5 h and the concentrations of IAM are (A) 10 mM, (B) 20 mM, (C) 40 mM and (D) 80 mM. | ||
What's more, there are three other groups of tryptic peptides of this kind which were named the group 2–4. And they also have been circled in Fig. 1. The mechanisms to produce these groups of peptides have not been clarified and the occurrence of these types of peptides will disturb the identification of the tryptic peptide which has the mutation site. Therefore it is necessary to use the TOF/TOF tandem mass spectrometry to explain the mechanisms of this phenomenon.
MS/MS analysis of tryptic peptide with mutations characteristic and its group
The Fig. 3 shows the MS/MS results of the TP1 and other peptides in the group 1, m/z 1951.9, m/z 2009.9 and m/z 2066.9. The fragment ions are b–y type ions and the y type ions are more abundant because of the C-terminal Arg or Lys which are more easily to be protonated. With plentiful fragment ions the amino acid sequence of m/z 1895.9 can be obtained to identify the mutation site. As the result, the second His from N-terminal of this peptide has changed to Ala, which conforms to the characteristic of H13A protein. It is also worth noticing that the peptide at m/z 1951.9 is the original tryptic peptide adding one acetyl group. In addition there are several extra fragment peaks from the MS/MS results of m/z 1951.9, such as m/z 713.2 (57 MW more than y6), m/z 828.3 (57 MW more than y7) etc. What's more, the fragment ion, m/z 713.2, has proved that the acetyl has added to y6 whose sequence is “AKPQGR”. But there is no fragment ion which has 57 MW more than y5, which refer to the fragment with sequence of “KPQGR”. This phenomenon indicates that the acetyl group has added to the Ala. In consideration of the methyl from side chain of Ala having difficulty to react with iodine acetamide, it is possible that the acetyl group firstly reacts to the carboxyl group on the side chain of Asp. When MS/MS occurred, the side chain carboxyl group preferred attacking the amido bond to produce anhydride as the previous literature reported (as shown in Fig. 4).1,33 Then the acetyl group transferred to the amino terminal of Ala to produce the fragment ion, m/z 713.2. | ||
| Fig. 3 MS/MS results of TP1 groups (a) TP1 peptide; (b) TP1 + 57 MW; (c) TP1 + 2 × 57 MW; (d) TP1 + 3 × 57 MW. | ||
 | ||
| Fig. 4 Proposed mechanism of acetyl group adding to Ala in the TP1 + 57 MW peptide. | ||
The MS/MS results of m/z 2010.1 and m/z 2067.1 are also shown in Fig. 3(c) and (d). According to the above mechanisms, these two ions declared that two or three acetyl groups can attached to the tryptic peptide. This phenomenon is different from the previous literature which proposed that the IAM just reacts with one kind of amino acids in one tryptic peptide.27 The characteristic ion at m/z 770.5 which are 2 × 57 MW higher than y6 illustrated that two acetyl groups can add to the fragment ion with sequence “AKPQGR”. Besides one acetyl group having been proved to add to the amino terminal of Ala, another most probable location site is the side chain amino of Lys. The new fragment ion, m/z 699.2 which doesn't exist in the MS/MS result of m/z 1951.9 can demonstrate the above assumptions. This ion is 2 × 57 MW higher than y5 whose sequence is “KPQGR”. And there is no fragment ion which has 57 or 2 × 57 MW more than the y4 with sequence “PQGR”. So the mechanism of fragment ion at m/z 699.2 is that one acetyl group can add to the Lys at its side chain amino and the other acetyl group would transfer from the amino terminal of Ala to the amino terminal of Lys during the Ala loss from the fragment ion “AKPQGR”.
In addition, the reactivity between amino acids and IAM has also been analyzed. Firstly from the analysis of y-type ions of TP1, it is obvious that IAM initially reacts with Asp in terms of the y9 ion as “AYDAKPQGR” and then another acetyl group binds to Lys as the appearance of the characteristic ion at m/z 770.5. So it is demonstrated that IAM is more readily to react with Asp than Lys. Secondly there is no fragment ion which is 3 × 57 MW higher than the y-type ions of peptide. But the intensity of ion group y13 at m/z 1494.7, y13 + 57 at m/z 1551.8 and y13 + 57 × 2 at m/z 1608.9 are the highest in the MS/MS result of TP1 + 57, TP1 + 57 × 2 and TP1 + 57 × 3 respectively which indicates that the y13 ion of TP1 can get two IAM. Additionally the third acetyl group is added to the b3 ion of TP1 with the appearance of b-type ion at m/z 459 (b3 + 57) (shown in ESI†). Combined with the MS/MS result of other tryptic peptides such as TP4 (see in Fig. 5), this binding site is more likely to be His. Since both the two part of the TP1 can react with IAM, it's necessary to distinguish which site can get alkylated more easily. From the MS/MS result of TP1 + 57, the fragment ion at m/z 713.2 (y6 + 57) indicates the first IAM binds to the Asp in terms of the “AYDAKPQGR”. However, the intensity of the y6 is still high enough demonstrating that other part of TP1 except “AYDAKPQGR” can also react with IAM and this binding site is proposed to be His as discussed above. And it is clear that the peak intensity of y6 is much higher than y6 + 57 which means His reacts easier than Asp when the TP1 getting monoalklated.
 | ||
| Fig. 5 MS/MS results of TP4 groups (a) TP4 peptide; (b) TP4 + 57 MW. | ||
MS/MS analysis of tryptic peptides of other groups
Other three groups of tryptic peptides which have multiple acetyl groups have also been investigated. Taking the group 4 with two fragment ions at m/z 2268.1 and m/z 2325.2 as an example, the MS/MS result firstly indicated that they stand for the same peptide with sequence “NVVVACENGLPVHLDQSIFR” locating in the C-terminal of protein (as shown in Fig. 5). After preparation, this peptide should add one acetyl group because of the Cys residue. When this peptide added another acetyl group to produce m/z 2325.2, the location site was possible in the side chain of His. And the reason is that there is the fragment ion, m/z 1072.7 which is 57 MW more than y8 with the sequence “HLDQSIFR”. But there is no fragment ion which has 57 MW more than y9 with the sequence “LDQSIFR”. Additionally when adding acetyl group, the molecular weight of peptide increased 57 MW but not 58 MW. So this acetyl group is possible adding to the secondary amine of His rather than tertiary amine to produce ionic formation. From the analysis of sequence “HLDQSIFR” above, we can find that IAM reacts with His in prior to Asp which matches the conclusion got from the analysis of TP1.The MS/MS results of the group 2 and 3 which have m/z 1314.7, m/z 1371.8, m/z 1428.8, m/z 1485.8 and m/z 1554.8, m/z 1611.8, m/z 1668.8 respectively are shown in Fig. S1 and S2.† The mechanisms of adding acetyl groups in these groups are determined as same as afore mentioned. The possible location site of acetyl group besides Cys is the side chain of Asp, Lys and His. From the result above, we proposed that multiple overalkylation reaction occurs in one tryptic peptide after protein digestion and the probable reaction site can be identified by MS/MS technique. And from the analysis of TP1 peptide group, the tendency of reaction between amino acids and IAM is Cys > His > Asp > Lys or N-terminal amine, which also different from previous literature.
In summary, four groups of tryptic peptides from H13A protein have been investigated by MALDI-TOF tandem mass spectrometry. The peptides in each groups successively increased by 57 MW. MS/MS results indicated that acetyl group can also react with other amino acids such as Asp, Lys and His.
Conclusion
In this work, the H13A protein which is the mutated angiogenin in the thirteenth position from N-terminal has been investigated using bottom-up mass spectrometry. Dithiothreitol and iodoacetamide have been used to open the disulfide bond and protect the thiol as the normal protocol before trypsin digested. When the digested protein was analyzed by MALDI-TOF mass spectrometry, a type of tryptic peptides which did not match the protein contains cleavage peptides by exact mass analysis has been found. These peptides have a special characteristic that having multiple 57 MW more than several protein contains peptides. Furthermore, MS/MS experiments have provided deeper understanding of the mechanisms of this phenomenon. Besides Cys, the excess IMA can also react other amino acids (e.g. Asp, Lys, His). This discovery can help us to exclude interference produced by iodoacetamide side effects and identify the peptides from digested protein accurately and rapidly using bottom-up method.Conflict of interest
The authors declare that they have no conflict of interest.Acknowledgements
We appreciate Professor Dr ZhengPing Xu from School of Medicine, Zhejiang University for his supporting K40I protein. We also appreciate the National Science Fund for Distinguished Young Scholars (No. 21025207), National Basic Research Program of China (No. 2011CB710800), Natural Science Foundation of China (No. 21505116) and the Scientific research funds of talents from Xuzhou medicine college, No. D2015014.References
- M. Guo, C. Guo and Y. Pan, Amino Acids, 2014, 46, 1939–1946 CrossRef CAS PubMed.
- M. Guo, J. Chen and Y. Pan, et al., PLoS One, 2014, 9, e104835 Search PubMed.
- C. Guo, L. Yue and Y. Pan, et al., J. Am. Soc. Mass Spectrom., 2013, 24, 381–387 CrossRef CAS PubMed.
- J. Fitzgerald, P. Kunnath and A. Walker, et al., Anal. Chem., 2010, 82, 4412–4419 CrossRef PubMed.
- S. Eshghi, S. Yang and X. Wang, ACS Chem. Biol., 2014, 9, 2149–2156 CrossRef PubMed.
- A. Grey, P. Chaurand and K. Schey, et al., J. Proteome Res., 2009, 8, 3278–3283 CrossRef CAS PubMed.
- K. Chan, P. Lanthier and J. Li, et al., Anal. Chim. Acta, 2009, 639, 57–61 CrossRef CAS PubMed.
- R. Aebersold and M. Mann, Nature, 2003, 422, 571–586 CrossRef PubMed.
- M. Dupré, S. Cantel and P. Verdié, et al., J. Am. Soc. Mass Spectrom., 2011, 22, 265–279 CrossRef PubMed.
- A. M. Frank and P. A. Pevzner, Anal. Chem., 2005, 77, 946–973 CrossRef.
- B. Paizs and S. Suhai, Mass Spectrom. Rev., 2005, 24, 508–548 CrossRef CAS PubMed.
- R. Boyd and A. Somogyi, J. Am. Soc. Mass Spectrom., 2010, 21, 1275–1278 CrossRef CAS PubMed.
- E. A. Kapp, F. Schutz and R. A. O'Hair, et al., Anal. Chem., 2003, 75, 6251–6264 CrossRef CAS PubMed.
- M. Baudet, P. Ortet and J. Armengaud, et al., Mol. Cell. Proteomics, 2010, 9, 415–426 CAS.
- N. Taouatas, M. M. Drugan and S. Mohammed, et al., J. Proteome Res., 2010, 9, 4282–4288 CrossRef CAS PubMed.
- L. S. Ferguson, F. Wulfert and S. Francese, et al., Analyst, 2012, 137, 4686–4692 RSC.
- R. Wang, K. Bao and C. N. Siu, et al., Analyst, 2013, 138, 6737–6741 RSC.
- J. Jiao, Y. Zhang and H. Lu, et al., Analyst, 2015, 140, 156–161 RSC.
- N. Rodthongkum, R. Ramireddy and W. V. Richard, et al., Analyst, 2012, 137, 1024–1030 RSC.
- L. Quinton, K. Demeure and R. Dobson, J. Proteome Res., 2007, 6, 3216–3223 CrossRef CAS PubMed.
- J. Hardouin, Mass Spectrom. Rev., 2007, 26, 672–682 CrossRef CAS PubMed.
- J. Pauleth, N. Solis and S. J. Cordwell, Biochim. Biophys. Acta, 2013, 1834, 372–379 CrossRef PubMed.
- Z. Yang and A. B. Attygalle, J. Mass Spectrom., 2007, 42, 233–243 CrossRef CAS PubMed.
- E. S. Boja and H. M. Fales, Anal. Chem., 2001, 73, 3576–3582 CrossRef CAS PubMed.
- Y. Shen, N. Tolic and R. D. Smith, et al., Anal. Chem., 2008, 80, 1871–1882 CrossRef CAS PubMed.
- D. M. Creasy and J. S. Cottrell, Proteomics, 2002, 2, 1426–1434 CrossRef CAS PubMed.
- J. Wang, X. Zhao, Y. Zhao, C. Ma, R. Zhong, X. Qian and W. Ying, Chin. J. Chromatogr., 2013, 31, 927–933 CrossRef CAS.
- X. Gao and Z. Xu, Acta Biochim. Biophys. Sin., 2008, 40, 619–624 CrossRef CAS PubMed.
- G. Cho, B. Kang and S. Kim, Mol. Cell. Biochem., 2010, 31, 8022–8029 Search PubMed.
- S. Alexandra, K. Matthew and M. Lsabela, J. Neurosci., 2012, 32, 5024–5038 CrossRef PubMed.
- J. Sebastia, D. Kieran and B. Breen, Cell Death Differ., 2009, 16, 1238–1247 CrossRef CAS PubMed.
- M. Guo, D. Tang and Y. Pan, et al., Anal. Chim. Acta, 2015, 865, 31–38 CrossRef CAS PubMed.
- C. G. Gu, G. Tsaprailis and V. H. Wysocki, Anal. Chem., 2000, 72, 5804–5813 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra18595e |
| ‡ These authors contributed to the work equally and should be regarded as co-first authors. |
| This journal is © The Royal Society of Chemistry 2015 |