DOI:
10.1039/C5RA18569F
(Paper)
RSC Adv., 2015,
5, 94191-94200
A novel chitosan/wheat gluten biofoam fabricated by spontaneous mixing and vacuum-drying†
Received
10th September 2015
, Accepted 26th October 2015
First published on 28th October 2015
Abstract
A new type of chitosan and wheat gluten biofoam is presented. The pore structure achieved relied solely on the specific mixing and phase distribution when a film was cast from an aqueous mixture of chitosan/wheat gluten solution, in the absence of any chemical blowing agent, porogen or expanding gas. The foam was obtained when the liquid phase was removed by vacuum drying, without the need for the traditional freeze-drying that is frequently used for pore formation. Soft foam samples could be prepared with stiffnesses from 0.3 to 1.2 MPa and a high rebound resilience (64 and 94% at compressive strains of 80 and 20%, respectively). The foams were relatively ductile and did not require any plasticiser to allow for in-plane deformation (20% compression) and smaller bending. Only open pores with sizes of the order of 70–80 μm were observed by microscopy. The density of all the foams was ∼50 kg m−3, due to the high porosity (96% air). The foams showed a rapid and large uptake of both non-polar (limonene) and polar (water) liquids. When immersed in these liquids for 1 second, the maximum uptake recorded was 40 times the initial mass of the foam for limonene and 8 times for water.
1. Introduction
Chitosan and wheat gluten (WG) have both attracted attention as alternatives to petroleum-based polymers. Chitosan-based porous materials have a potential for use in separation filters for waste-water treatment and protein separation,1,2 in scaffolds for wound dressings,3,4 in tissue engineering,5–7 and as templates for porous ceramics.8 WG are important in food due to its good foaming properties.9–11 Recently, WG foams were explored as a potential alternative for controlled release devices or for indoor use as sound insulation materials.12–15 Only a few reports, however, have been published on chitosan/WG solid films.16,17
It is known that these polymers can be used to prepare foams/porous structures. Porous chitosan materials have been prepared using a phase inversion in which an inorganic material, such as sodium chloride or silica,1,2 or a hydrophilic polymer (polyethylene glycol)18 was used as porogen in order to produce pores in chitosan. Gas foaming is another process used to obtain chitosan foams.4,6,19 The pores were made by bubble-forming mechanical agitation of the chitosan solution, or by gas injection into an aqueous solution. Since the bubbles must persist until the material has dried, a technique to increase the viscosity of the liquid is required, such as alkali treatment or the use of crosslinking agents or surfactants. WG foams have also been made with emulsifiers or fluid carbon dioxide or by drying the solution at a temperature above 45 °C.12,13 Among many methods to create the foam of the dissolved materials, freeze-drying has been frequently used to preserve the porosity in both chitosan and WG foams.3,5,7,8,14,15,20,21 The basic procedure for freeze-drying is that an aqueous solution of the polymer is first frozen at a low temperature, after which a porous material is obtained by sublimation of ice under vacuum. Recently, a more advanced technique, ice-segregation-induced self-assembly (ISISA), was used to make chitosan-based foams with more complex structures.22,23
In the present study, we present a novel method to prepare biobased foams. The method involves mixing chitosan and WG solutions, and it requires no chemical blowing agent, porogen, injected gas or a freezing liquid phase. The foam preparation method has a narrow processing window. The initial separate phases (observed as several cm large domains) in the mixture gradually changed without any agitation to a new phase distribution where phase separation was still observed, but on a smaller scale. After this “spontaneous” mixing, the foams were obtained by vacuum drying. The samples with foam structures were characterized with respect to pore structure, porosity, sorption kinetics, and mechanical properties.
2. Experimental
2.1. Materials
Commercial WG powder was kindly supplied by Reppe AB, Lidköping, Sweden. According to the supplier, the gluten protein content (according to Mod NMKL nr 6, Kjeltec, Nx5.7) was 77.7% and the starch content was 5.8% (Ewers, polarimetric method). The chitosan provided by Sigma Aldrich had a ![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) w = 790 kDa and n = 210 kDa. The degree of deacetylation, as revealed by infrared spectroscopy, was 76%.24 Anhydrous acetic acid (purity = 98%) and sodium sulfite (purity = 98%) were obtained from Sigma Aldrich. R(+) Limonene was obtained from Alfa Aesar (purity = 97%).
w = 790 kDa and n = 210 kDa. The degree of deacetylation, as revealed by infrared spectroscopy, was 76%.24 Anhydrous acetic acid (purity = 98%) and sodium sulfite (purity = 98%) were obtained from Sigma Aldrich. R(+) Limonene was obtained from Alfa Aesar (purity = 97%).
2.2. Preparation of foams
Fig. 1 presents the experimental scheme. The chitosan acetic acid solution (CS) and the WG solution (WGS) were prepared according to a method presented by Chen et al.16 Chitosan powder was dissolved in aqueous acetic acid (0.05 M) to obtain solutions with concentrations of 0.5, 1, 1.5 and 2 g chitosan per 100 mL of solvent. These solutions are referred to as 0.5, 1, 1.5 and 2 wt%, respectively. The solutions were stirred overnight (800–1000 rpm for all stirring operations) and the pH was then 4.0 ± 0.2. Water, combined with a reducing agent (sodium sulfite), was used as a solvent for WG. Water and 0.3 wt% of sodium sulfite, based on dry WG, were mixed together and stirred for 15–20 min, and the WG powder was then added slowly to the solution. After 30–40 min stirring, the pH was lowered to 4 by the addition of acetic acid. This solution was then stirred for 30–40 min. The concentration of WG was 0.5, 1, 1.5, and 2 g per 100 mL of solution (0.5, 1, 1.5 and 2 wt%).
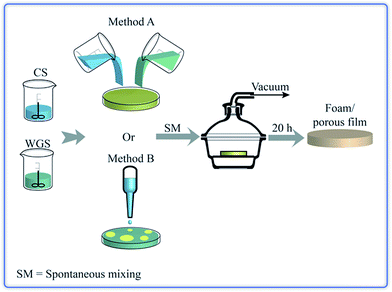 |
| | Fig. 1 The methodology to produce chitosan/WG blend foams. | |
After the two solutions had been filtered using a TexWipe TX309 cloth (118 × 60 threads per inch, pore size: 100–200 μm), the CS and WGS were poured together into a polystyrene Petri dish. The mixtures of CS and WGS were designated e.g. 1C/1W, which corresponded to a mixture of the solutions of 1 wt% CS and 1 wt% WG. Ten combinations were tested: xC/1W, 1C/xW and xC/xW where x was 0.5, 1, 1.5 and 2. In order to keep the number of combinations low, the mass fractions of pure chitosan and WG was always 1/1 in the liquid mixtures. In addition, for each combination of CS and WGS, the total mass of the liquid mixture in the dish was kept at 10, 16 or 22 g. After CS and WGS were added together, the dishes were kept at 20 ± 1 °C for 20–40 min. The mixing of the components was monitored using a NIKON-D40 camera. The dishes were subsequently placed in a desiccator connected to a SCANVAC coolsafe™ 100-9 PRO freeze dryer equipped with an EDWARDS-RV3 vacuum pump (pressure: 0.1–0.5 hPa; temperature of the cold trap: −96 ± 1 °C), without allowing the samples to freeze. Dried films were obtained after 20–24 h of vacuum treatment. The films were stored in a desiccator with silica drying agent for at least 24 h before being further studied.
Pouring the CS into the WGS was referred to as method A (Fig. 1), although pouring WGS into CS instead did not affect the final film structure. Method A was applied for all the mixture combinations, except for the 0.5C/1W mixture. In this case, the porous content varied from batch to batch and, in order to achieve a high extent of foaming in the sample, the CS was added dropwise to WGS. This is referred to as method B (Fig. 1).
2.3. Foam characterization
2.3.1. Solution viscosities. A Brookfield Cap 2000+ viscometer, calibrated with a viscosity standard (CAP0L) of 56.1 cP, was used to assess the dynamic viscosities (ηd) of the solutions. Because forced mixing occurs when the cone spindle of the viscometer is spinning, well-blended mixtures of CS and WGS, obtained by vigorous stirring with a magnetic stirrer, were prepared and used in the viscometer. The rotation velocity and measuring time were set to 500 rpm and 45 s, respectively. A higher rotation velocity made it difficult to measure the most dilute chitosan solution and a lower rotation velocity increased the risk of having inaccurate data. In addition, the rotation velocity was in the range where the viscosity was essentially independent of the shear rate.25 An average dynamic viscosity for each mixture combination was calculated from at least 3 replicates. Because the viscosity of pure acetic acid (1.14 cP) and water (1 cP)26 were close to each other under ambient conditions, the acetic acid/water viscosities were assumed to be 1 cP.
2.3.2. Field-emission scanning electron microscopy (FE-SEM). The morphology of the foams was examined in a FE-SEM (Hitachi S-4800, Hitachi Science System, Ltd., Japan). Film specimens frozen in liquid nitrogen were broken and their fracture surfaces were examined. The samples were coated with platinum in a Denton Vacuum chamber, using an Agar High resolution Sputter Coater (208RH), equipped with a platinum target/agar thickness controller. Because of the different morphologies at the two foam surfaces, the surface exposed to the vacuum drying was referred to as the top surface, while the surface touching the Petri dish was referred to as the bottom surface. The sizes of the pores were obtained from the FE-SEM images by measuring the diameter in random directions of at least 160 pores. The samples used for the assessment of pore diameter were cut from central parts of the foams.
2.3.3. Density and porosity of foams. A Mettler Toledo balance (AL104, reading accuracy = 0.1 mg), equipped with a density determination kit, was used for the density measurements according to the Archimedes principle. The density of the solid films (ρ) was calculated according to:| |
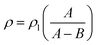 | (1) |
where ρl is the density of the n-hexane (laboratory reagent grade, Fisher Scientific) liquid; A is the weight of the sample in air and B is the weight of the sample in the n-hexane liquid. Since the solid films were prepared from the same formulations as the corresponding foams, although with a different loading (16 g instead of 22 g), the densities (ρ) reported for the solid films in Table 2 were taken to be the solid-phase (bulk) densities (ρb) of the foams. Apparent densities (ρa) of the foams were obtained by assessing the mass and volume of the foam samples, which were cut into cylindrical specimens with a diameter of 14 mm using a cork borer. A digital caliper ruler (Absolute AOS Digimatic, Mitutoyo, Japan) was used to measure the size of the samples, and the average thickness and diameter was used in the volume calculation.
Table 1 Extent of foaming in the sample (%)a
| CS (wt%) |
WGS (wt%) |
| 0.5 |
1 |
1.5 |
2 |
| To simplify the calculations, the circular film was divided into four quarters and the extent of the foaming was, based on these four quarters, 0, 25, 50, 75 and 100%. |
| 0.5 |
100 |
100 |
— |
— |
| 1 |
25 |
100 |
75 |
50 |
| 1.5 |
— |
25 |
75 |
— |
| 2 |
— |
0 |
— |
0 |
Table 2 Foam structure
| Combination |
Thickness (μm) |
Bulk densitya (kg m−3) |
Apparent densityb (kg m−3) |
Pore size (μm) |
Porosity (%) |
| Density of the solid material using the Archimedes principle (16 g solutions). Density of the foams. |
| 0.5C/0.5W |
409 ± 83 |
1207 ± 60 |
46 ± 5 |
75.9 ± 35.6 |
96.1 ± 0.4 |
| 0.5C/1W |
468 ± 110 |
1303 ± 59 |
52 ± 6 |
70.3 ± 39.8 |
96.0 ± 0.4 |
| 1C/1W |
1054 ± 140 |
1328 ± 20 |
54 ± 6 |
81.6 ± 37.7 |
95.9 ± 0.5 |
The porosity (P) was determined from the ratio of the apparent density (ρa) to the bulk density (ρb) of the samples, calculated as in eqn (1):
| |
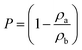 | (2) |
2.3.4. Liquid uptake by the foams. The initial mass of the foam samples was measured on a Precisa, XR 205SM-DR balance. Samples were immersed in limonene or water for 1 s and 1 min. The wet sample mass was recorded within 10 s after withdrawing the sample. Because the foams disintegrated on immersion for 1 min in water, no data for 1 min water uptake were obtained.
2.3.5. Infrared (IR) spectroscopy. The top and bottom surfaces of the foam samples were characterized by IR spectroscopy (Perkin-Elmer Spectrum 2000, Perkin-Elmer Inc., USA.) equipped with a single reflection ATR accessory (Golden Gate from Specac Ltd., Kent, England). A 4 cm−1 wavelength resolution and 32 scans were used for all the spectra taken. At least 3 locations on the different surfaces of each foam sample were analysed.
2.3.6. Compression tests. Cylindrical specimens with a diameter of 14 mm were cut from the foam samples using a cork borer. The specimens were conditioned for 48 h at 23 ± 1 °C and 50 ± 2% RH before testing, and the specimens were tested at 23 ± 1 °C and 50 ± 2% RH in an Instron 5944 universal testing machine with a 500 N load cell. The compression rate was 1 mm (min)−1 as suggested by Liu et al.7,27 The maximum strain was set to either 20 or 80%. The rebound resilience (R) was determined according to.28–30| |
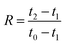 | (3) |
where t0, t1, and t2 are the thicknesses before loading, during loading (20 or 80% strain) and after unloading, respectively. The thicknesses before and after loading were determined using a digital caliper ruler (Absolute AOS Digimatic, Mitutoyo, Japan), and the thickness during the loading was calculated from the original thickness at the applied strain. The number of replicates was 5.
2.3.7. Tensile testing. Dumb-bell-shaped specimens were punched out from the samples using a sample die (ISO 37, type 3), providing specimens where the narrow section had a width of 4 mm and a length of 16 mm. The specimens were conditioned for at least 48 h at 23 ± 1 °C and 50 ± 2% RH before testing. The thickness was measured on each specimen at 5 locations in the test section and the average values were used to calculate the cross-sectional area and stress. The specimens were tested in the environment in which they were conditioned using an Instron 5944 universal testing machine with a crosshead speed of 2.5 mm min−1 and a 500 N load cell. All values are averages of at least 5 replicates.
3. Results and discussion
3.1. Preparation of foams
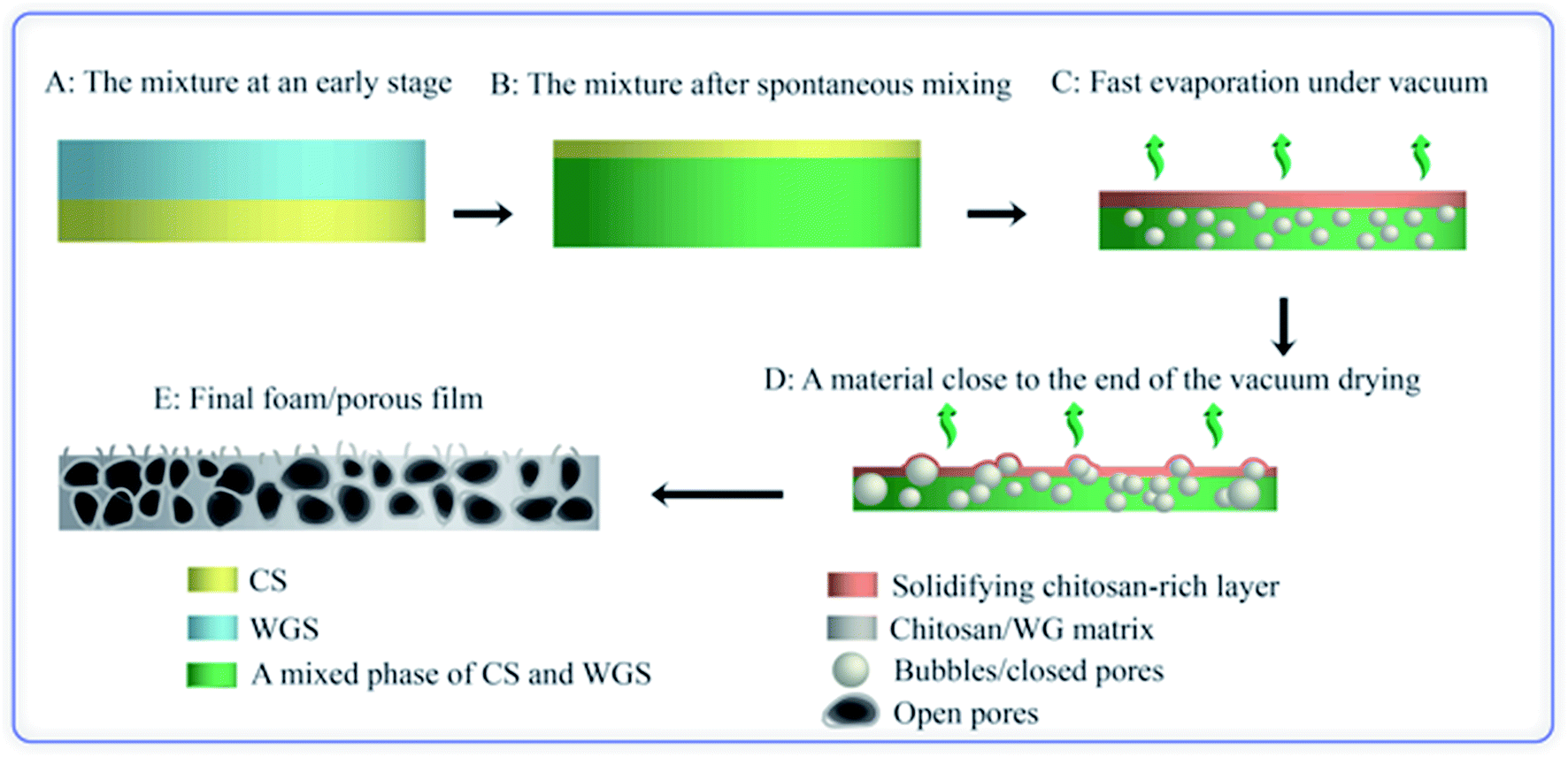
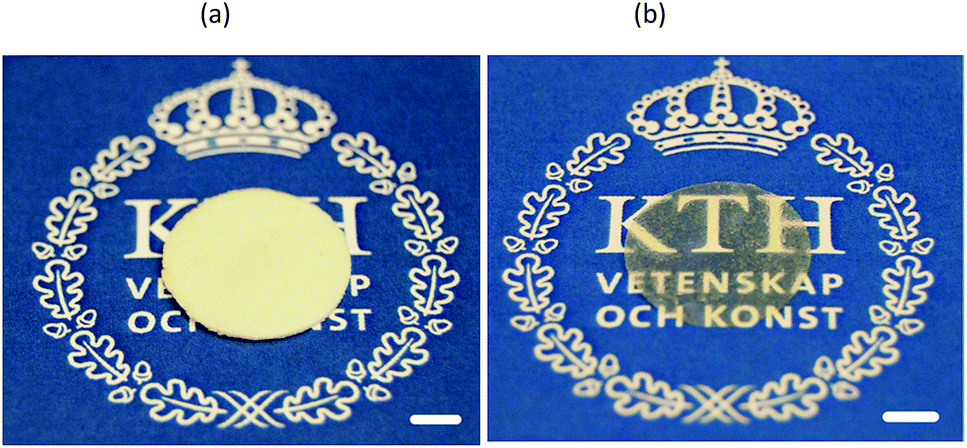
3.1.1. Initial experiments. In the previous investigation on solid chitosan/WG films, it was decided to increase the rate of film formation by using vacuum drying.16 Surprisingly, it was observed that a few films became porous during the drying, although no chemical blowing agents or porogen had been added and freeze-drying was not used. It was however difficult to repeat these experiments. Only a few films formed porous structures and the porosity was not even uniformly distributed through the film samples. SEM images revealed that there were large oval holes at the top surface whereas cracks were observed on the bottom surfaces (not shown). The cross-section showed a sandwich structure in which the top and bottom surfaces layers appeared to be denser than the middle section. This heterogeneous structure indicated a layering of the chitosan and WG blend. With increasing magnification, small particles with a size of ca. 1–3 μm were observed in the sample. The preliminary studies suggested that the preferred phase structure to obtain porous samples was biphasic on different scales. Indeed, by allowing a poor mixing, as outlined in Fig. 1 (method A), it was possible to fully reproduce the foams (Fig. 2(a)).
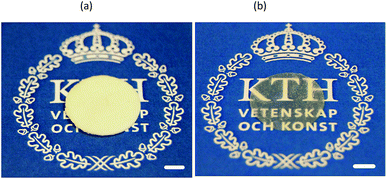 |
| | Fig. 2 (a) A foamed and (b) solid film prepared from 1C/1W. The scale bar is 0.5 cm long. | |
3.1.2. Different combinations of CS and WGS. After the initial experiments, method A (Fig. 1) was used with a number of CS/WGS combinations to make foams. For each formulation, a total mass of 10, 16 or 22 g was decanted into a Petri dish.These amounts were chosen because (a) 22 g was the maximum mass that fitted into the Petri dish and (b) a lower mass (16 and 10 g) yielded information as to whether the formation of the foam depended on the total mass of the solution. In fact, none of the 10 g mixtures formed any porous structure and only a few films dried from the 16 g mixtures were porous. None of these samples were however fully porous. The 22 g mixtures gave samples with a dominantly porous structure, although not all the 22 g mixtures formed fully foamed films. Since the foamed regions were white and the solid regions transparent/translucent it was convenient to assess the foam content visually. Table 1 shows the extent of foaming in the sample as a function of the CS/WGS content. 100% porous samples were obtained only from the 0.5C/0.5W, 0.5C/1W and 1C/1W mixtures, and the effects on the porous structure of varying the polymer concentrations in the CS and WGS, and of varying the CS/WGS ratio, were investigated. The total mass of the liquid mixture and the mass ratio of chitosan and to WG were kept constant, so that varying the concentration of either CS or WGS resulted in a change in the total polymer content in the mixture. For 1C/xW and xC/1W (x = 0.5, 1, 1.5 and 2), the total polymer concentrations in the mixtures were 0.7, 1, 1.2, and 1.3 wt%. For the xC/xW mixtures, the total polymer concentrations were 0.5, 1, 1.5 and 2 wt%.
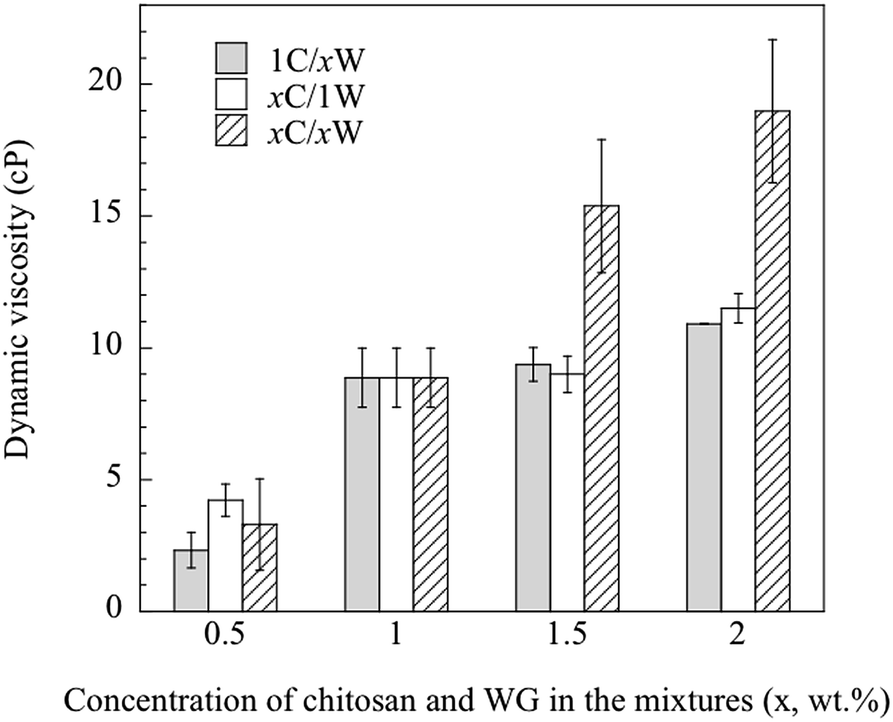
3.1.3. Viscosity of the solutions. Besides the total polymer content in the mixture, the viscosity of the CS was assumed to affect significantly the development of the porous structure. The dynamic viscosity of the CS increased significantly with increasing chitosan concentration (from ca. 20 cP at 0.5 wt% to ca. 120 cP at 2 wt%, Fig. S1, ESI†). The 2 wt% CS had a gel-like appearance. On the other hand, the viscosity of the WGS was, within this WG concentration range, too low to be measured with the viscometer available. The viscosity has however been reported earlier (1.95–2.10 cP at a pH of 3.5–5) for a 1 wt% WGS.11 The dynamic viscosities of the mixtures are presented in Fig. 3. Overall, the viscosity of the mixtures increased with increasing polymer content. If the mixtures were miscible, it should be possible to calculate the blend viscosity by the rule of mixtures, i.e. from the separate viscosity contributions of CS and WGS to the total blend viscosity:31| |
ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ηblend = lnηCSωCS + lnηWGSωWGS ηblend = lnηCSωCS + lnηWGSωWGS
| (4) |
where ηCS and ηWGS are the viscosities of CS and WGS and ωCS and ωWGS are the relative mass fractions.
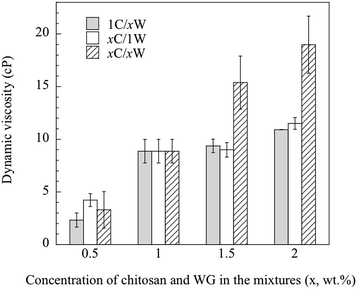 |
| | Fig. 3 Dynamic viscosity of the different mixtures. | |
Any deviations from eqn (4) were considered to be due to immiscibility of the two solutions and their polymers (chitosan and WG). This was tested by the following procedure. Knowing the viscosity of 1C/xW and CS with 1 wt% chitosan, the viscosity of WGS was calculated using eqn (4) for the WG content of x = 0.5–2 wt%. The WGS and CS viscosities were then used to calculate the viscosity of the xC/1W mixtures, and the calculated values were compared with the experimental xC/1W viscosities.
Fig. 4 shows that the predicted viscosities (eqn (4)) were almost constant, whereas the experimental viscosity increased with increasing the concentration of CS. The poor prediction of the experimental values indicated that the well-blended mixtures were immiscible even though they all appeared homogenous on a gross scale. Hence, it was important to study step by step how the foam was fabricated from these immiscible mixtures.
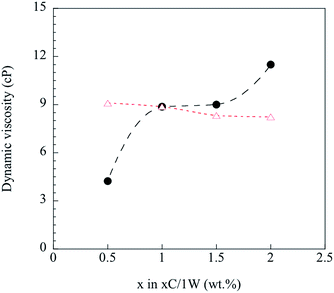 |
| | Fig. 4 Calculated ( ) and measured ( ) and measured ( ) dynamic viscosity. ) dynamic viscosity. | |
3.1.4. Spontaneous mixing. Fig. 5 shows how the phase distribution of the mixture of CS and WGS changed with time. This mixing procedure was referred to as spontaneous mixing. At the instant when the CS was poured into the WGS (1C/1W), a two-phase structure was observed; the CS being optically clear whereas the WGS was opaque. From above it was observed that the clear coarse two-phase structure gradually disappeared. The side view revealed that the WGS-rich phase was initially located above the CS-rich phase (4 min image in Fig. 5). With time, the CS and WG gradually mixed, but a thin transparent layer of CS formed at the upper surface. The last top-view image shows that mixing was complete after 40 min. A similar spontaneous mixing was observed for 0.5C/0.5W and 0.5C/1C, but the mixing of 0.5C/0.5W was three times faster than for 1C/1W (Fig. S2(a)†), and that for 0.5C/1W (not shown) was slightly faster than for 1C/1W. On the other hand, the 2C/2W sample, which did not form any porous structure, did not show this spontaneous mixing (Fig. S2(b)†). The observed spontaneous mixing behaviour in both the vertical and horizontal directions and the formation of a CS layer at the top surface thus seem to be necessary in order to obtain a porous structure. In other words, phase separation on a gross scale has to be attained. Forced mixing/stirring yielded a visually uniform structure, but these samples did not form a foamed structure on vacuum drying. Spontaneous mixing did not occur in the mixture with 2 wt% of CS due to the high viscosity of CS (Fig. S1†), and none of these mixtures yielded a porous structure.
 |
| | Fig. 5 Images of the mixing of WGS and CS (1C/1W) taken (a) from the top and (b) from the side. The extension of the WGS into the CS is marked by black arrows, and the final thin CS-rich layer is marked by a white arrow. | |
3.1.5. Effects of vacuum drying on the structure the solid material. For the blends undergoing spontaneous mixing, foam was formed only when the mixtures were dried in vacuum. The temperature inside the desiccator containing the samples and connected to the dryer was 14 °C in the first few hours and ca. 20 °C at the end of the treatment, which means that the liquid phase did not freeze during the vacuum drying. The effects of the forced drying in vacuum on the structures of the blend are illustrated by FE-SEM. Our previous study showed that a film cast from a rapidly stirred solution of CS and WGS was essentially an immiscible system with WG particles, having an average diameter of 1.3 μm, uniformly dispersed in the chitosan-rich matrix.16 Fig. 6 shows cross-sections of the 1C/1W films obtained by drying under ambient conditions and in vacuum. These films did not form a porous structure in vacuum due to insufficient mass of the mixture added to the dish. In the former case, several large dark regions were seen randomly distributed along the bottom part of the solid film (Fig. 6(a)). At a higher magnification, it was observed that these regions contained agglomerates of many irregular particles, probably rich in WG. The dark region spread over a larger volume when the solid film was dried more rapidly in vacuum (Fig. 8(b)). It is possible that these differences played a role in whether the solution became a solid or a porous material at the 22 g loading. In addition to the agglomerates, well-dispersed WG particles with diameters of 1–4 μm were found in the continuous chitosan-rich matrix in the solid film, regardless of the drying conditions.
 |
| | Fig. 6 FE-SEM micrographs of cross-sections of solid films (prepared from the 16 g mixture) of 1C/1W dried at ambient conditions (a) and under vacuum (b). Arrows point at WG-rich agglomerates. The inset figure is a magnification of a part of an agglomeration. | |
 |
| | Fig. 7 FE-SEM micrographs of (a) the top surface and (b) the bottom surface of a foam sample prepared from the 1C/1W mixture. The white arrow points to the base of a crater with holes and the black arrows point to WG-rich domains. | |
 |
| | Fig. 8 (a) FE-SEM micrograph of the cross-section of the porous 1C/1W foam, the shapes of the pores being indicated by crossed arrows. The white arrow points at the top surface skin. (b) Cross-section of the 0.5C/1W foam with a top surface of marked residual parts of broken bubbles (highlighted in the inset figure). (c) Images of the 1C/1W foam taken from the region close to the bottom (1), further from the bottom (2 and 3) and close to the top (4). Black arrows mark wheat-gluten-rich regions and white arrows mark chitosan-rich regions. The scale bars are 50 μm long. | |
3.2. Morphology of foams and porous structure formation
Fig. 7 shows surfaces of a foamed film prepared from the 1C/1W mixture (22 g). The other two formulations (0.5C/1W and the 0.5C/0.5W) showed a similar morphology (not shown). The foam structure of the top surface differed distinctly from that of the bottom surface. The top surface showed many crater-like holes (diameter ∼ 0.4 mm), whereas the bottom surface showed cracks and smaller holes. Besides the different surface structures, different colour patterns were observed on the top and bottom surfaces. Dark spots were observed on the bottom surface whereas the top surface was brighter and more homogeneous in colour. The dark spots were probably WG-rich domains (Section 3.1).
The cross-section of a foam sample showed that the pores were irregular and open (Fig. 8(a)). The structure of the cross-section was different from that of the top and bottom surfaces. The top surface resembled a “skin” that covered the porous interior and contained bubbles/closed pores or broken bubbles (craters/wall fragments) (Fig. 8(a) and (b)). The pores close to the bottom surface were less elongated than those close to the top surface (Fig. 8(a)). A closer look revealed that the colour of the pore walls changed in the vertical direction, from darker and more heterogeneous WG-rich regions (bottom) to brighter and smoother chitosan-rich regions (top) (Fig. 8(c)). Small WG particles with diameters of 1–3 μm were observed in the chitosan-rich regions, but these became gradually less pronounced from the bottom towards the top (Fig. S3†). The distribution of WG and chitosan in the vertical direction indicated that the final state of the spontaneous inter-mixing was preserved in the foam after vacuum drying.
Based on these observations, the following mechanism is suggested for the development of the porous structure, seen in Fig. 9. During the spontaneous mixing, the initial completely separated CS and WGS phases are mixed horizontally and vertically. The final phase distribution of the mixture is a mixed WGS and CS phase (probably biphasic) under a thin CS “layer”. There is a gradient in WG in the mixed phase, and more WG in the bottom region. The final phase distribution is preserved until the mixture has dried in vacuum. As the liquid evaporates in the vacuum drying, the mixture becomes concentrated and starts solidifying. Due to the good foaming property of WGS and increasing viscosity of CS at increasing concentration, the viscous/solid chitosan-rich top layer prevents most of the small bubbles formed in the mixed phase from growing too large and breaking and collapsing. Furthermore, the CS in the mixed phase also protects the pores from collapsing, even though the pore walls may break at the later stage (leading to an open pore structure). The surface layer also forms bubbles, which eventually break and form a crater-like structure. Holes at the bottom of the crater are vents where the last liquid phase evaporated (Fig. 7(a)). The final foam is a combination of these polymers and their properties.
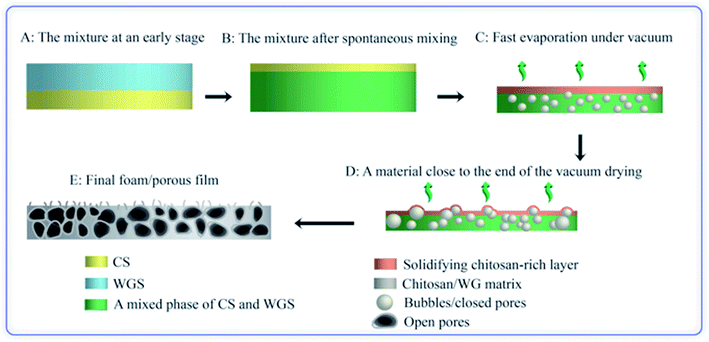 |
| | Fig. 9 Illustration of the porous structure/foam fabrication. | |
3.3. IR spectroscopy
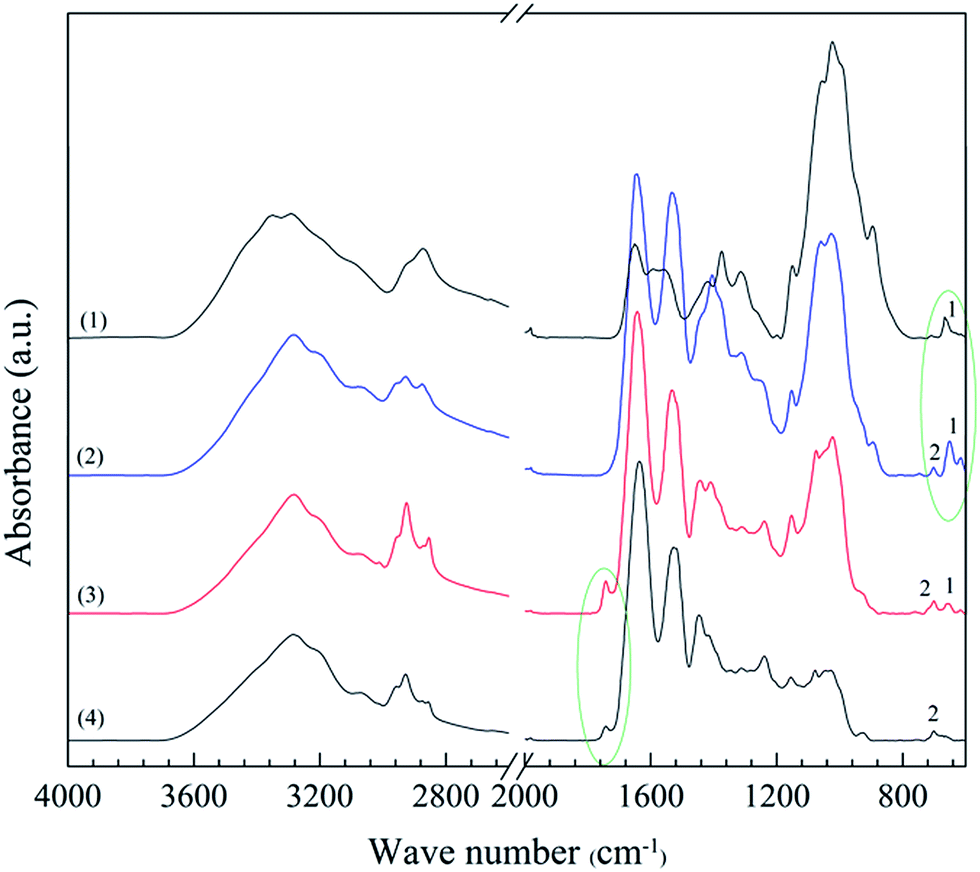
The top and bottom surfaces of the porous samples were characterized by IR spectroscopy (Fig. 10). The thickness of the surface penetrated by IR depends on the wavelength and is typically 1–5 μm for a solid polymer.32 The penetration is probably deeper in porous materials, however still being a small fraction of the total thickness. A few IR bands could be assigned exclusively to either the chitosan or WG and made it possible to detect their presence. The 670 cm−1 band (band 1 in Fig. 10), assigned to the out-of-plane bending vibration associated with –NH–,33 was prominent in the spectra of the top surface of the porous samples, which indicated that the top surface material was rich in chitosan. The 1743 cm−1 band, due to carbonyl-containing species in WG, in the spectra of the bottom surface confirmed that WG-rich regions dominated at the bottom of the foam samples. Furthermore, band 2 at 702 cm−1 (Fig. 10) was presented only in the spectra of the top and bottom surfaces of the blend foam and in the spectrum of pristine wheat gluten, suggesting that it was a characteristic peak of wheat gluten. The ratio of the intensities of bands 1 and 2 depended on the wheat gluten content. The chitosan and wheat gluten contents at the top and bottom surfaces were obtained from this ratio. The results showed 20 and 56 wt% of wheat gluten in the top and bottom surface layers, respectively. Even the solid films (collapsed foams) had more wheat gluten in the lower parts.
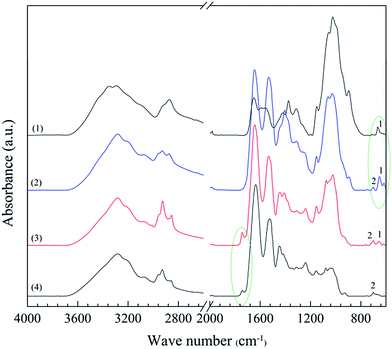 |
| | Fig. 10 IR spectra of (1) chitosan powder, (2) the top surface of a 1C/1W foam, (3) the bottom surface of the foam and (4) WG powder. | |
3.4. Dimensions of the foams
The thicknesses of the foam with a fully porous structure are given in Table 2. The thickness of the foams (1054 ± 140 μm) were 15 times thicker than the solid blend films (68 ± 19 μm) prepared from the same formulation with stirring. The thickness increased with increasing polymer content in the mixtures (cf. 0.5C/0.5C, 0.5C/1W and 1C/1W films). The bulk densities of the foam samples increased with increasing polymer concentration in the solution, indicating that the degree of molecular packing increased as the film dried from a more concentrated polymer solution, or alternatively that voids were present in the films dried from the more dilute polymer solutions (0.5C/0.5W, 0.5C/1W). It was impossible to verify the existence of such voids because of the heterogeneous cross-sectional structure. Due to this polymer-concentration/density relationship, the apparent density of the foams, which was quite low, increased with increasing polymer concentration. The porosity was higher (96%, Table 2) than that reported for chitosan-based2,6,7,18,20,27 and WG-based15,21 foams. The average pore size (ca. 70–80 μm, Table 2) of the present foams was similar to that reported for WG-based foams15,21 and within the same range as that reported for chitosan-based porous materials (20–350 μm).1,4–6
3.5. Liquid uptake in the foams
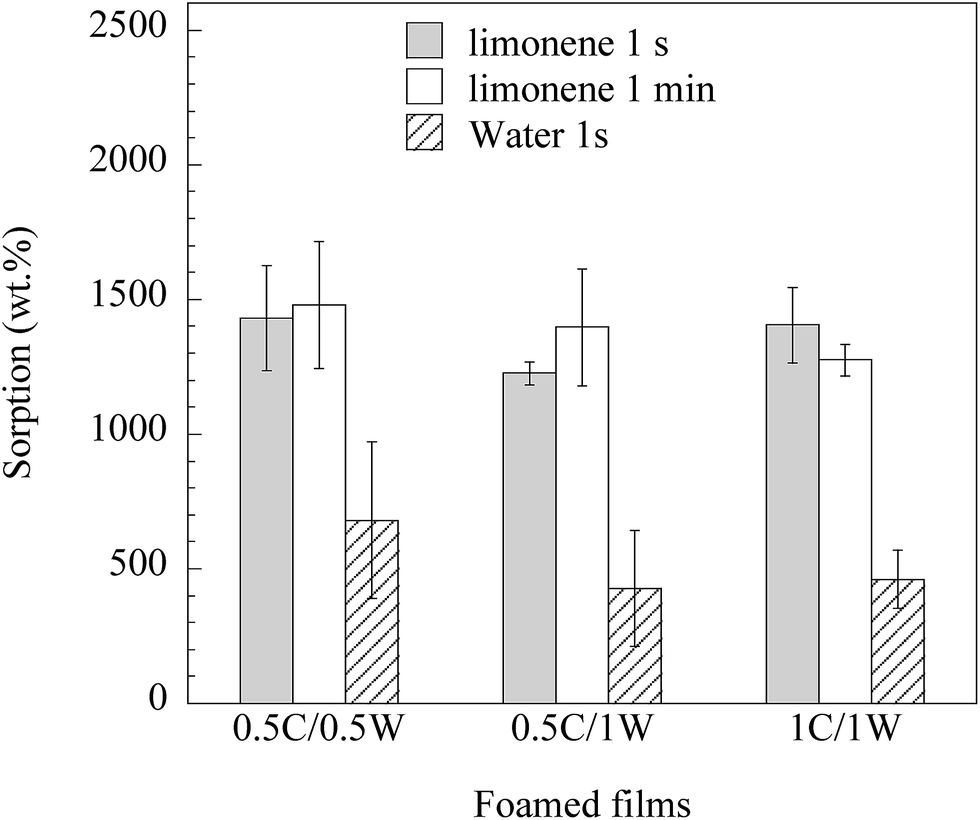
Fig. 11 shows that the foams rapidly sorbed a large amount of limonene (and less, but still a large amount, of water). The foam prepared from the 1C/1W formulation had 15 times the mass of the dry film after 1 s immersion in limonene. The rapid sorption of limonene in the first second was a consequence of the presence of mainly open pores. In a previous study on freeze-dried WG foams,15 the uptake of limonene in WG foams was only ca. 1 times the initial mass after immersion for three seconds. The possible reasons for the lower uptake in the freeze-dried foams than in the present foams were the lower porosity and less open pores in the former. The average water uptake in 1 s in the present foams ranged between 4 and 7 times the initial dry weight. In contrast to limonene, which essentially only filled the pores, water also entered the pore walls. This weakened the porous structure and all the films disintegrated within 1 min during immersion. Porous materials based on chitosan have been reported to sorb 18 times their initial weight of a 0.9% NaCl aqueous solution (simulated body fluid).4 Similar uptakes have also been reported using a buffer solution (PBS) with pH = 7.4.7,19,27 In all these cases, however, the uptake was measured after at least 30 min immersion in the liquid.
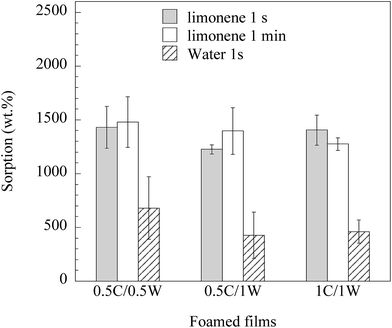 |
| | Fig. 11 Uptake in foams immersed in limonene and water for different times. | |
It was surprising that the limonene uptake was faster than the water uptake, considering that the foams were hydrophilic. However, we have also observed this phenomenon in the case of freeze-dried WG foams. In order to study what is causing this behaviour, the wetting behaviour of the two liquids were compared. The shape of a limonene and water droplet placed on a solid 1C/1W film surface was investigated (with a foam it would not be possible to investigate the spreading/wetting behaviour due to the very heterogeneous surface). The limonene droplet spread rapidly over the film surface whereas the water droplet spread more slowly and not to the same extent as the limonene droplet. This is a consequence of that water has a higher surface tension than limonene34,35 and also that water, in contrast to limonene, penetrated into the solid film. Evidently, the differences in surface tension (lower tension liquid fills the foam cells more rapidly) and penetration behaviour led to a slower uptake of water than of limonene (their viscosities are similar26,36). Because of the extensive swelling of the solid material, water was however expected to reach a significantly higher final uptake than limonene, provided the foam samples could be kept mechanically intact during a longer exposure.
Samples from the 0.5C/0.5W and 0.5C/1W formulations grew, occasionally, to a thickness of ca. 2 mm during the vacuum drying and their maximum uptake during 1 s of limonene and water was 40 and 8 times the initial dry weight, respectively. These large values are presented separately from those in Fig. 11, since we currently have no methodology to reproduce these highly-extended foams to 100%. However, the results indicate that the liquid uptake may be increased even more than that presented in Fig. 11, if additional optimization can be achieved.
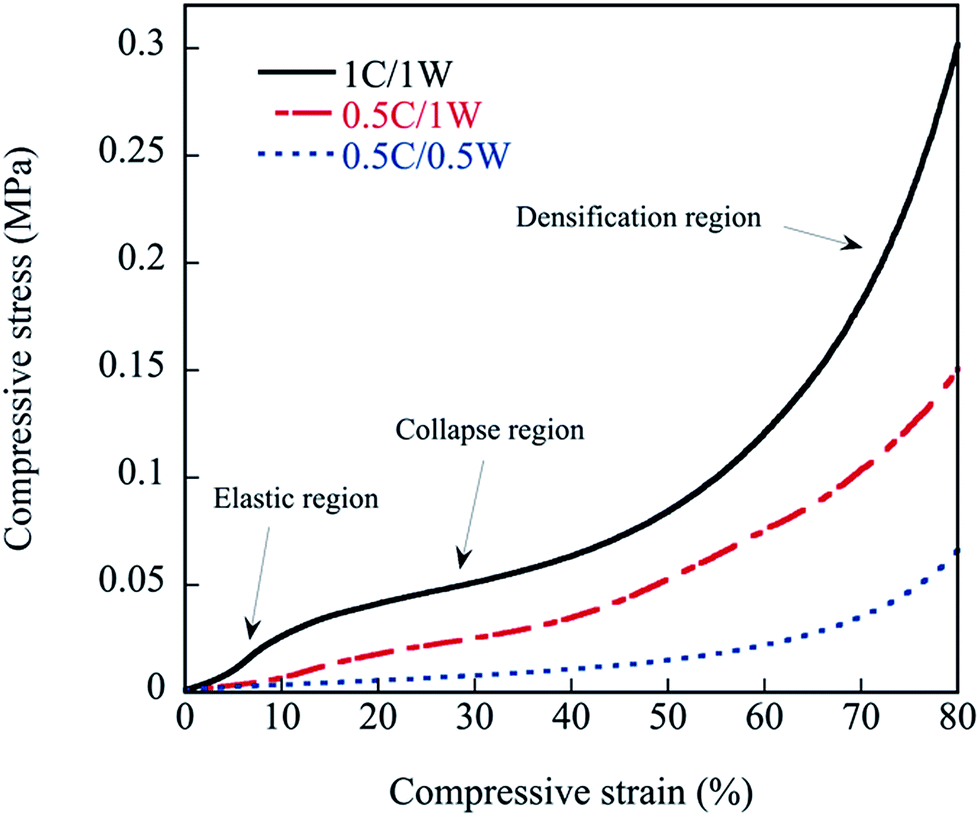
3.6. Compressive properties of foams
The behaviour of the foams under compression is presented in Fig. 12. As is commonly observed with foams, three regions appeared in the stress–strain curve of the 1C/1W film. The first region corresponds to an essentially elastic deformation where the stress increased essentially linearly with the strain. In the second collapse region, the pore walls broke/collapsed, and this was followed by a third densification region involving the deformation and compaction of the solid material. It was more difficult to separate the three regions in the stress–strain curves of 0.5C/1W and 0.5C/0.5W (Fig. 12).
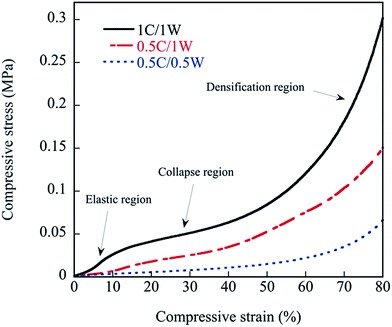 |
| | Fig. 12 Compressive stress–strain curves of foamed samples. | |
The mechanical properties of the foam samples are summarized in Table 3. The compression modulus and compressive strength increased with increasing polymer concentration in the solutions (0.5–1 wt%, Table 3). This may be related to the increase in the bulk density of the foams with increasing polymer concentration in the mixture. It has been reported previously that the tensile modulus increases with increasing concentration of chitosan in the solution, and it has been suggested that this is due to concentration-dependent chitosan molecular conformations and packing during film formation.37 The moduli presented in Table 3 were a hundred times greater than that of a bio-scaffold composed of chitosan and gelatin.7 As a complement tensile properties of the solid films were measured (Table 3). These were made in the same way as the foams but without the vacuum drying. In addition, films were made by first stirring the two solutions together for 30 min at ambient conditions before the casting, a technique used in ref. 16. The tensile modulus, as well as the tensile stress at break, was insignificantly different when comparing the three solid films, with and without the stirring. The only noticeable trend was that the scatter in the tensile stress at break was higher for the films obtained without stirring the solution. Hence, there were no clear correlation between the trends in the compression data among the foams and the tensile data of the films. Even though there was always a phase separation in all systems, it is interesting to note that the absolute values of the tensile stiffness and strength were similar with and without the stirring before casting. In fact, the tensile strength of the solid films here were close to that of pure chitosan.16 A two-phase/phase-separated system does not necessarily imply poor mechanical properties, as shown by Ceseracciu et al.38 on new types of biodegradable elastomers based on corn starch and polydimethylsiloxane. The rebound resilience was as high as ∼90% after 20% deformation, indicating a large degree of elastic deformation. It is interesting to note that, despite the absence of a plasticizer, the presence of chitosan made the foams significantly more flexible than solid films of pure WG, and than films of chitosan/WG.16
Table 3 Compression data
| Formulation |
Compression modulus (MPa) |
Compressive stressa (MPa) |
Tensile modulusb (MPa) |
Tensile modulusc (MPa) |
Tensile stress at breakb (MPa) |
Tensile stress at breakc (MPa) |
Rebound resilienced (%) |
Rebound resiliencee (%) |
| At a strain of 80%. Solid films without stirring. Solid films with stirring. The resilience was assessed after a strain of 80%. Resilience assessed after a strain of 20%. |
| 0.5C/0.5W |
0.3 ± 0.03 |
0.1 ± 0.01 |
3580 ± 130 |
3710 ± 290 |
28.6 ± 9.1 |
33.9 ± 4.4 |
64 ± 13 |
94 ± 8 |
| 0.5C/1W |
0.6 ± 0.1 |
0.2 ± 0.04 |
3610 ± 218 |
3250 ± 200 |
41.1 ± 4.8 |
36.4 ± 3.2 |
41 ± 5 |
92 ± 12 |
| 1C/1W |
1.2 ± 0.2 |
0.3 ± 0.05 |
3300 ± 480 |
3490 ± 480 |
31.4 ± 12.2 |
43.6 ± 6.3 |
39 ± 5 |
89 ± 10 |
Although the differences were often within the standard deviation, a monotonous decrease in rebound resilience with increasing polymer concentration in the mixture was observed.
4. Conclusions
It has been shown that it is possible to make renewable and biodegradable foams/porous films from a blend of two essentially incompatible polymers (chitosan and WG) by spontaneous mixing and vacuum drying. The mainly open pore structure was formed without the need for any chemical blowing agent, porogen, freeze-drying or injected gas. The density was in the same low range as that of conventional polymer foams. The foams had a large and rapid sorption/uptake capacity for both polar and non-polar liquids. In the case of water, the film disintegrated/degraded when subjected to a long immersion time, which is advantageous in a sanitation as well as a recycling perspective. The films were soft with a high rebound resilience at moderate compressive strains. In addition, they were tough enough to withstand mild mechanical treatment without the need of a plasticiser. By combining chitosan and WG, one makes use of the toughness, clarity and antimicrobial properties of chitosan and the good foaming properties and lower price of WG. Applications of the present foamed films include the use of them as “soft flexing surfaces” and ad-/absorbants.
Acknowledgements
The Chinese Scholarship Council and KTH are thanked for providing financial support for Fei Chen.
References
- A. C. Chao, S. H. Yu and G. S. Chuang, J. Membr. Sci., 2006, 280, 163–174 CrossRef CAS.
- X. Zeng and E. Ruckenstein, Ind. Eng. Chem. Res., 1996, 35, 4169–4175 CrossRef CAS.
- C. Stefanescu, W. H. Daly and I. I. Negulescu, Carbohydr. Polym., 2012, 87, 435–443 CrossRef CAS.
- C. Z. Bueno and A. M. Moraes, J. Appl. Polym. Sci., 2011, 122, 624–631 CrossRef CAS.
- H. Liu, K. Nakagawa, D. Chaudhary, Y. Asakuma and M. O. Tadé, Chem. Eng. Res. Des., 2011, 89, 2356–2364 CrossRef CAS.
- S. K. Nandi, B. Kundu and D. Basu, Mater. Sci. Eng., C, 2013, 33, 1267–1275 CrossRef CAS PubMed.
- Y. Liu, M. An, L. Wang and H. Qiu, J. Macromol. Sci., Part B: Phys., 2014, 53, 309–325 CrossRef CAS.
- J. D. Mathias, N. Tessier-Doyen and P. Michaud, Int. J. Mol. Sci., 2011, 12, 1175–1186 CrossRef CAS PubMed.
- J. E. Kinsella, Food Chem., 1981, 7, 273–288 CrossRef CAS.
- B. G. Thewissen, I. Celus, K. Brijs and J. A. Delcour, J. Agric. Food Chem., 2011, 59, 1370–1375 CrossRef CAS PubMed.
- T. Mita, K. Nikai, T. Hiraoka, S. Matsuo and H. Matsumoto, J. Colloid Interface Sci., 1977, 59, 172–178 CrossRef CAS.
- L. Andreani, R. Cercená, B. G. Z. Ramos and V. Soldi, Mater. Sci. Eng., C, 2009, 29, 524–531 CrossRef CAS.
- S. Quester, M. Dahesh and R. Strey, Colloid Polym. Sci., 2014, 292, 2385–2389 CAS.
- T. O. J. Blomfeldt, R. Kuktaite, T. S. Plivelic, F. Rasheed, E. Johansson and M. S. Hedenqvist, RSC Adv., 2012, 2, 6617–6627 RSC.
- T. O. J. Blomfeldt, R. T. Olsson, M. Menon, D. Plackett, E. Johansson and M. S. Hedenqvist, Macromol. Mater. Eng., 2010, 295, 796–801 CrossRef CAS.
- F. Chen, X. Monnier, M. Gällstedt, U. W. Gedde and M. S. Hedenqvist, Eur. Polym. J., 2014, 60, 186–197 CrossRef CAS.
- S. K. Park and D. H. Bae, Food Sci. Biotechnol., 2006, 15, 133–137 Search PubMed.
- M. Zeng, Z. Fang and C. Xu, J. Appl. Polym. Sci., 2004, 91, 2840–2847 CrossRef CAS.
- A. Jagadeesh, G. G. Chaudhari, D. K. Bal, S. Patra and S. Ganguly, Int. J. Polym. Mater. Polym. Biomater., 2015, 64, 134–139 CrossRef CAS.
- Z. Y. Gu, P. H. Xue and W. J. Li, Polym. Adv. Technol., 2001, 12, 665–669 CrossRef CAS.
- Q. Wu, R. L. Andersson, T. Holgate, E. Johansson, U. W. Gedde, R. T. Olsson and M. S. Hedenqvist, J. Mater. Chem. A, 2014, 2, 20996–21009 CAS.
- J. Liu, Z. He, J. Xue and T. T. Y. Tan, J. Mater. Chem. B, 2014, 2, 2478–2482 RSC.
- Z. He, J. Liu, Y. Qiao, C. M. Li and T. T. Y. Tan, Nano Lett., 2012, 12, 4738–4741 CrossRef CAS PubMed.
- M. R. Kasaai, Carbohydr. Polym., 2008, 71, 497–508 CrossRef CAS.
- J. K. Hwang and H. H. Shin, Korea Aust. Rheol. J., 2000, 12, 175–179 Search PubMed.
- J. G. Bleazard, T. F. Sun and A. S. Teja, Int. J. Thermophys., 1996, 17, 111–125 CrossRef CAS.
- Y. Liu, M. An, H. X. Qiu and L. Wang, Polym.-Plast. Technol. Eng., 2013, 52, 1154–1159 CrossRef CAS.
- X. Dong, G. Sui, J. Liu, A. Guo, S. Ren, M. Wang and H. Du, Compos. Sci. Technol., 2014, 100, 92–98 CrossRef CAS.
- S. Shen, Y. Zhao, H. Du, M. Wang, L. Zhao, G. Tian and J. Liu, Ceram. Int., 2014, 40, 8905–8909 CrossRef CAS.
- X. Dong, H. Lv, G. Sui, J. Liu, A. Guo, X. Tao, X. Xu and H. Du, Mater. Sci. Eng., A, 2015, 635, 43–49 CrossRef CAS.
- L. A. Utracki, in Encyclopedia of polymer blends: processing, ed. A. I. Isayev, WILEY, 2011, vol. 2, pp. 27–30 Search PubMed.
- M. S. Hedenqvist, M. Krook and U. W. Gedde, Polymer, 2002, 43, 3061–3068 CrossRef CAS.
- S. Mima, M. Miya, R. Iwamoto and S. Yoshikawa, J. Appl. Polym. Sci., 1983, 28, 1909–1917 CrossRef CAS.
- N. B. Vargaftik, B. N. Volkov and L. D. Voljak, J. Phys. Chem. Ref. Data, 1983, 12, 817–820 CrossRef CAS.
- R. L. Shubkin, B. F. Kanegsberg and E. Kanegsberg, in Synthetics, Mineral Oils, and Bio-based Lubricants, ed. L. R. Rudnick, Taylor & Francis Group, 2nd edn, 2012, p. 728 Search PubMed.
- R. A. Clara, A. C. Gomez Marigliano and H. N. Solimo, J. Chem. Eng. Data, 2009, 59, 1087–1090 CrossRef.
- C. A. Kienzle-Sterzer, D. Rodriguez-Sanchez and C. Rha, Makromol. Chem., 1982, 183, 1353–1359 CrossRef CAS.
- L. Ceseracciu, J. A. Heredia-Guerrero, S. Dante, A. Athanassiou and I. S. Bayer, ACS Appl. Mater. Interfaces, 2015, 7, 3742–3743 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra18569f |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence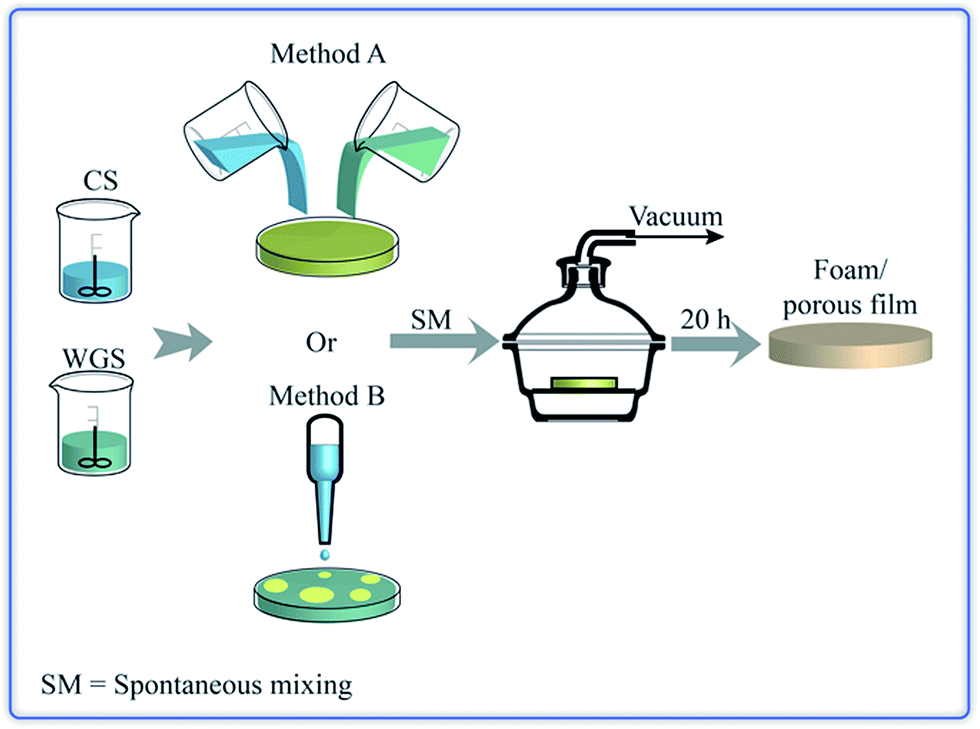
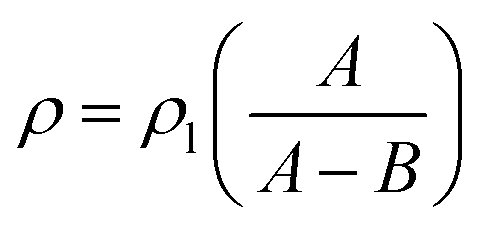
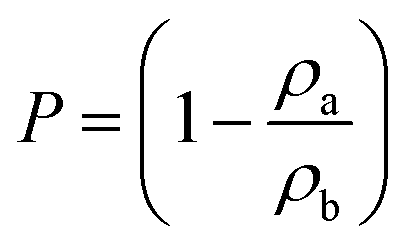
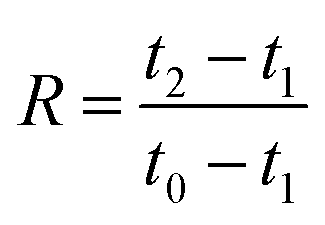
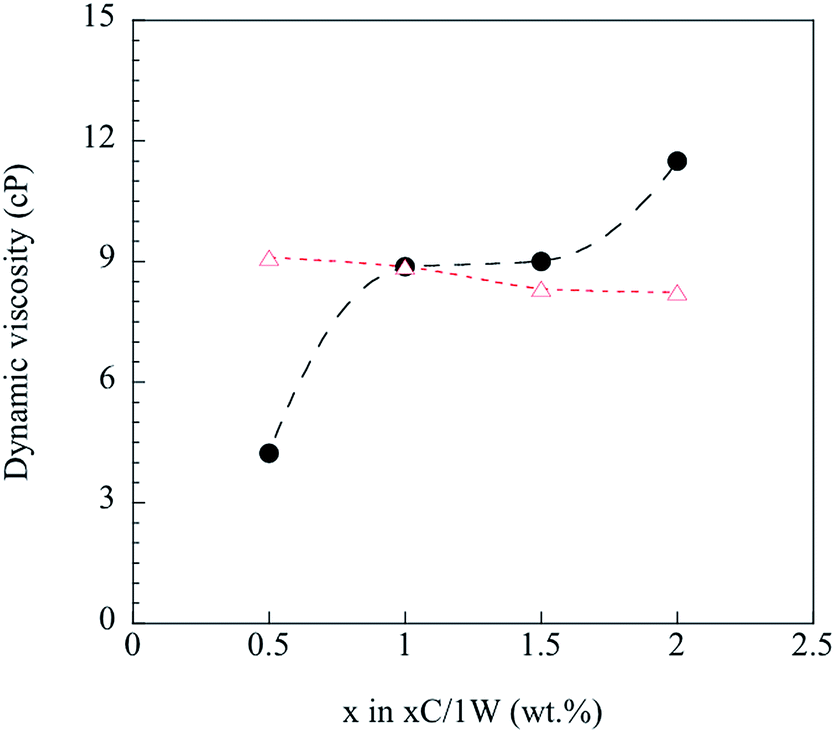
 ) and measured (
) and measured ( ) dynamic viscosity.
) dynamic viscosity.