
Kinetics of hydrazine reduction of thin films of graphene oxide and the determination of activation energy by the measurement of electrical conductivity
Abstract
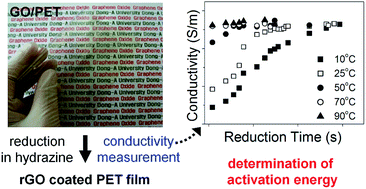
We fabricated transparent conductive gas barrier films by reducing graphene oxide (GO) coatings on polyethylene terephthalate (PET) films. GO solutions were cast on pure PET films, and the film surfaces were chemically reduced in a hydrazine hydrate solution. By measurement of the electrical conductivity of the films, the chemical reduction kinetics at the surface were examined as a function of reduction time, reductant concentration, and temperature. Based on these measurements, we determined the activation energy, 16 kcal mol−1, which corresponds to the barrier for the anchoring of the hydrazine either opposite to an epoxide ring or at a top site adjacent to a surface OH group. When the GO film was reduced to a carbon-to-oxygen (C/O) atomic ratio of 4.7, the electrical conductivity was 2.0 × 103 S m−1, with a light transmittance of 67% at 550 nm. We also revealed that the oxygen permeability of the film is a single exponential function of C/O atomic ratio. The reduced GO (rGO)/PET films exhibited an oxygen permeability of 7.97 × 10−8 Barrer (1 Barrer = 10−10 cm3 cm cm−2 s−1 cm Hg−1), providing optically transparent and electrically conductive polymer films for gas barrier applications.
 Please wait while we load your content...
Please wait while we load your content...

