
Synthesis and characterization of pyrimidine-containing hexaarylbenzene derivatives†
Dengqing Zhang*a,
Gang Wanga,
Rong Lia,
Xianying Li*b,
Yunjie Xianga,
Zhen Zhanga and
Wusong Jin*a
aCollege of Chemistry, Chemical Engineering and Biotechnology, Donghua University, 2999 North Renmin Road, Songjiang, Shanghai 201620, P. R. China. E-mail: dqzhang@dhu.edu.cn; wsjin@dhu.edu.cn
bSchool of Environmental Science and Engineering, Donghua University, 2999 North Renmin Road, Songjiang, Shanghai 201620, P. R. China. E-mail: seanlee@dhu.edu.cn
First published on 16th November 2015
Abstract
Three pyrimidine-containing claw-shaped unusual hexaarylbenzene derivatives have been successfully synthesized by cobalt-catalyzed cyclotrimerization of corresponding 3,3′-diaryldiphenylacetylenes. Cyclotrimerization of symmetric tolan derivative 6 gave symmetric product 1, while unsymmetric tolan derivative 15 afforded two geometric isomers, C3-symmetric 2 and unsymmetric analogue 3, which have been successfully separated by preparative thin-layer chromatography in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratios. The coordination of 1 with Ag+, Cu2+ and Zn2+ was studied by UV-vis, fluorescence spectra and TEM.
:2 ratios. The coordination of 1 with Ag+, Cu2+ and Zn2+ was studied by UV-vis, fluorescence spectra and TEM.
Introduction
Hexaarylbenzene derivatives (HABs) have attracted considerable attention due to their potential application in broad fields such as graphitic materials,1 self-assembly,2 energy storage,3 aromatic hydrocarbons,4 energy transfer,5 charge delocalization,6 and organic zeolites.7 So far, a variety of HABs with different functional groups have been synthesized,8 and their electrochemical and optical properties have been intensively characterized.9 Most of reported HABs were focused on introducing the functional groups into para-position of peripheral phenyls to form star-shaped geometry, however, the molecules substituted in meta-position of peripheral phenyls were still limited despite the fact that these unusual HABs might show different optical, electronic, self-aggregation and coordination properties.On the other hand, introduction of azaheteroaryl groups such as pyridyl, pyrimidinyl and imidazolyl etc. into HABs skeleton opened the possibility to coordinate with various metal ions, which hardly realized by full phenyl groups substituted HABs. As a heteroaryl moiety, pyrimidinyl modified HABs was least exploited although pyrimidine has high electron affinity and good coplanarity acting as an appropriate building block in construction of semiconductors,10 liquid crystalline materials,11 electroluminescent diodes,12 and fluorescent molecules.13 Moreover, pyrimidine can participate in hydrogen bonding formation and also can coordinate with various metal ions.14 Draper and co-workers have reported the synthesis of N-heterosuperbenzene by cyclodehydrogenation of pyrimidinyl-contained hexaarylbenzene,15 and its transition metal complexes have shown interesting optical, electrochemical and structural properties.16 However, up to now, the synthesis and characterization of pyrimidine-contained hexaarylbenzenes has rarely been reported. In this work, we designed and synthesized three kinds of claw-shaped unusual hexaarylbenzenes, where the pyrimidyl groups meta-substituted at peripheral phenyls, and investigated their coordinating ability with metal ions (Chart 1).
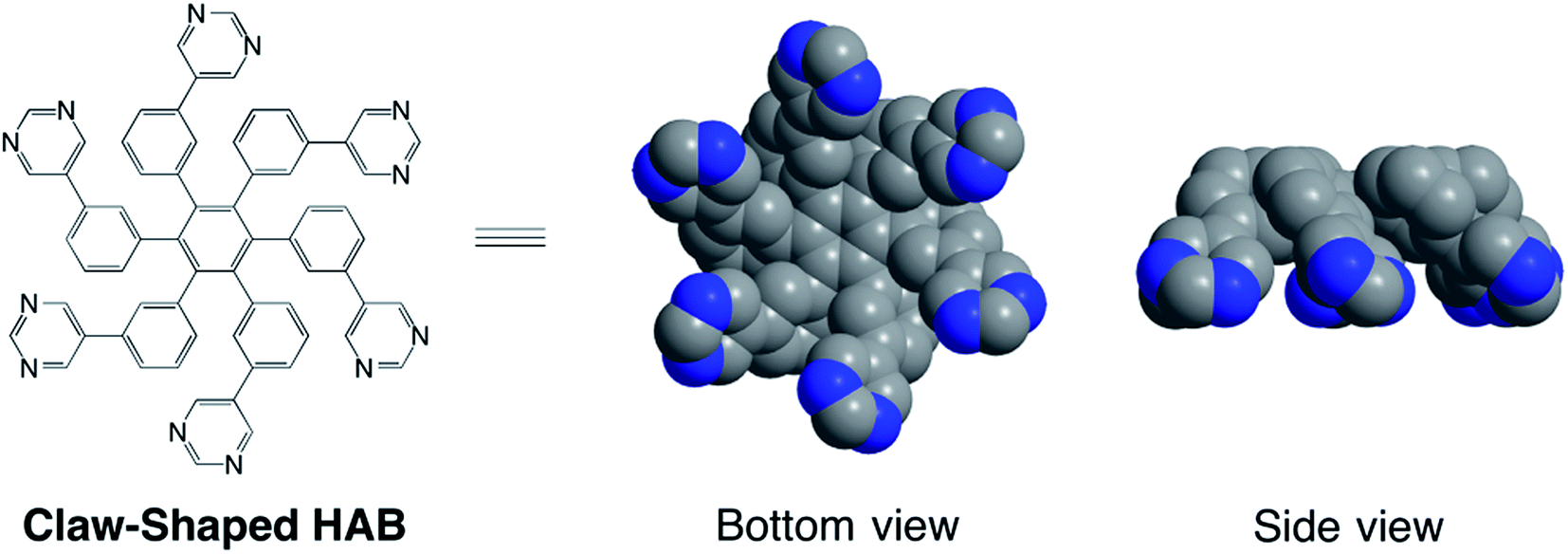 | ||
| Chart 1 Molecular structure and space-filling modes of claw-shaped meta-substituted hexaarylbenzene. | ||
Results and discussion
The symmetric 3,3′-disubstituted tolan derivative 6 was obtained in 75% yield by Suzuki coupling reaction (Scheme 1). Highly symmetric hexaarylbenzene 1 with six pyrimidine groups was synthesized by cobalt-catalyzed cyclotrimerization of the corresponding tolan precursor 6 in 90% yield (Scheme 1). Although other metal-complexes have also been known to catalyze this type of reaction, the cobalt complex is the most effective.17 | ||
| Scheme 1 Synthesis of HAB derivative 1. | ||
As shown in Fig. 1a, the 1H NMR spectrum of 1 exhibited two types of aromatic signals at downfield. Signals around at δ = 8.40–8.29 ppm integrating for 12 hydrogen atoms were assignable to the hydrogen atoms of the pyrimidine rings. Another one from 7.25 to 6.93 ppm was characterized to the aromatic protons of the phenyls. The multiplet rather than singlet of proton signals of pyrimidyl in compound 1 at room temperature revealed that six pyrimidyl rings were not magnetic equivalence. In general, six phenyl groups of HABs were arranged around a central benzene ring in a propeller-like fashion, and the dihedral angle between the phenyl substituent and the central benzene ring was approximate 60° which reduced direct orbital overlap (ortho–ortho interactions) and conjugational effects.18 In the present work, the pyrimidine pendants located at meta-position of peripheral phenyl rings, the terminal pyrimidine rings and peripheral phenyl rings were nonplanar due to the ortho–ortho interactions, which might cause the molecule adopting claw-shaped configuration rather than two-dimentional star-shaped geometry (Chart 1). Molecules with this configuration further self-aggregated by strongly π–π interactions between deficient electron pyrimidyl and phenyl ring of the neighboring molecule to form oligomers, which resulted in multiplet signals in 1H NMR spectra. When temperature was increased to 50 °C, the proton signals of pyrimidyl tended to decreased to two peaks and shifted to downfield (Fig. 1b), which implied the dissociation of oligomers accelerated at higher temperature. Moreover, the molecular structure of 1 was also characterized by MALDI-TOF mass spectrometry, the desired molecular ion-peak and isotopic distributions was observed (Fig. 2).
 | ||
| Fig. 1 Partial 1H NMR spectra of 1 in aromatic region (400 MHz, CDCl3). | ||
 | ||
| Fig. 2 MALDI-TOF mass spectrum of 1. | ||
For comparison, compounds 2 and 3 were synthesized over seven steps (Scheme 2). Firstly, compound 8 was synthesized from 7 according to a literature procedure,19 then 8 was converted to 9 by Suzuki coupling reaction. The TMS group of 9 was easily replaced by iodine using ICl as an iodination reagent. Next, compound 12, which obtained by reaction of 11 with 3-bromoiodobenzene, reacted with trimethylsilylacetylene (TMSA) yielded 13. Deprotection of 13 with tetrabutylammonium fluoride (TBAF) gave 14 in quantitative yields. The resulted terminal acetylene derivative 14 was then coupled with 10 via Sonogashira cross coupling to afford 15. Finally, the C3-symmetric 2 and unsymmetric analogue 3 were synthesized by cyclotrimerization of unsymmetric tolan derivative 15.
 | ||
| Scheme 2 The synthetic route to HAB derivatives 2 and 3. | ||
In principle, cobalt-catalyzed cyclotrimerization of tolan derivatives following the metallacyclopentadiene intermediates mechanism,18,20 the alkynyl π-complex (a) was firstly formed from two equivalents of alkyne with cobalt, and then two metallacycles (b) and four alkynyl π-complexes (c) were produced as intermediates, and then seven membered metallacycloheptatrienes (d) were formed, the resulting isomers were produced by reductive elimination reaction (Fig. 3). Therefore, two geometric isomers 2 and 3 were formed by cyclotrimerization of unsymmetric 15 (Scheme 2). The separation of isomers was very difficult by column chromatography. However, after many trials, we successfully separated the isomers by preparative thin layer chromatography with mixture of ethyl acetate/dichloromethane/petroleum ether (1:10:10) as eluent in 27% of 2 and 56% of 3 in yield, respectively (Fig. S20, Rf,2 = 0.90 and Rf,3 = 0.65; see ESI.† The polarity of C3-symmetric 2 was smaller than that of unsymmetric analogue 3). The ratio of 2 to 3 was not in agreement with that of statistical results because the steric and electronic factors might play important role in deciding the regiochemical outcome of the reaction.21 By means of MALDI-TOF mass spectrometry measurement, molecular ion-peaks appeared at 2007.92 and 2007.83 for 2 and 3 respectively (Fig. S17 and S19, see ESI†), which were identical to their theoretical molecular weight.
 | ||
| Fig. 3 Catalytic cycle for the cyclotrimerization of unsymmetric alkyne. | ||
The pyrimidine-containing hexaarylbenzene derivatives 2 and 3 did not exhibit the aggregation phenomena although their structures were similar to 1. In the 1H NMR spectrum of 1, two types of aromatic signals for C3-symmetric 2 or unsymmetric analogue 3 at downfield was observed (Fig. 4). Multiplet signals around δ = 7.19–6.87 ppm for both 2 and 3 were assigned to protons of phenyls, however, different peak patterns were clarified that peak split for unsymmetric 3 was more complicate, which proved that two isomers were completely separated. Interestingly, proton signal of the pyrimidine in 2 or 3 appeared only as singlet at δ = 8.39 ppm in 1H NMR spectra at room temperature (Fig. 4), which was quite different from that of 1. As mentioned above, the self-aggregated 1 possessing six deficient electron pyrimidyl rings exhibited the multiplet signals in 1H NMR spectrum (Fig. 1). Nevertheless, only three pyrimidyl rings for both 2 and 3 were insufficient to induce the self-aggregation between molecules. Therefore, they existed as monomer rather than oligomer in solution, if any, that dynamically, which caused singlet signal of pyrimidyl rings of C3-symmetric 2. As to unsymmetric 3, singlet signal might be owed to that chemical shift values of three pyrimidyl were very closed and overlapped together. In fact, no change of their peak patterns and chemical shifts were observed in 1H NMR spectra even at high temperature. These results indicated that the number of pyrimidyl groups in HABs play an important role for self-aggregation.
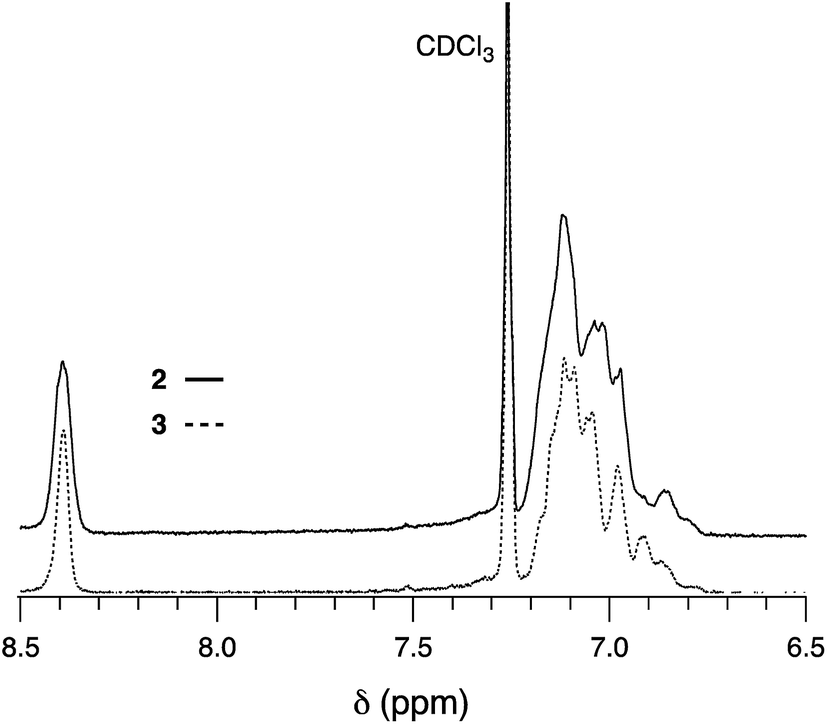 | ||
| Fig. 4 Partial 1H NMR spectra of 2 and 3 in aromatic region (400 MHz, CDCl3). | ||
Pyridine or pyrimidine-containing compounds were widely used as ligands in coordination chemistry for generating supramolecular objects with various morphologies, such as rotor, sandwich, nanoball and MOFs.22 Newly synthesized claw-shaped hexaarylbenzenes with pyrimidine units, which were expected to coordinate with metal ions under an appropriate condition to form amusing nanostructure. Therefore, the coordination with metal ions was investigated using 1 as a platform. Fig. 5 shows the absorption and fluorescence spectral changes of 1 as a function of Ag+, Cu2+ and Zn2+ in CH2Cl2 solution at room temperature. The UV-vis spectrum of 1 showed the maximum band at 255 nm, which was attributed to overlapping π–π* excitations.23 Upon addition of AgCF3SO3 into the solution of 1, the absorption band at 255 nm decreased remarkably, and concomitantly a new band at 237 nm appeared (Fig. 5a). Complexation also took place immediately when Cu2+ or Zn2+ was added into the system. As shown in Fig. 5a, the 255 nm absorption band decreased following the formation of new band centered at 242 nm. However, the interesting issue is the entirely different fluorescence spectra changes after addition of Ag+, Cu2+ and Zn2+ (Fig. 5b). Free 1 exhibited a structureless fluorescence emission at 382 nm (λex = 255 nm), upon addition of Ag+, the λmax em undergone a blue shift to 337 nm. Although no significant difference in absorption spectroscopy after addition of Cu2+ and Zn2+, obvious changes were observed in fluorescence spectroscopy. When Cu2+ was added to the solution of 1, the emission intensity quenched, while the emission intensity greatly increased along with red-shift by 110 nm when Zn2+ was introduced (Fig. 5b). Further mechanism research, particularly to prove the origin of the changes, are in progress.
 | ||
| Fig. 5 UV-vis and fluorescence spectra (excitation at 255 nm) of 1, 1 + Ag+, 1 + Cu2+ and 1 + Zn2+ in dichloromethane (1 × 10−5 mol L−1) at room temperature. | ||
To enable direct visualization of the solid state morphologies resulting from molecular interactions between metal ions and 1, transmission electronic microscopy (TEM) was employed. As indicated by Fig. 6a, molecules of metal-free 1 self-aggregated into sphere nanostructures with different sizes. Highly monodispersed silver nanoparticles were coated on the sphere nanostructures of 1 when Ag+ was added (Fig. 6b), the nanospheres with silver nanoparticles had more uniform sizes than that of metal-free 1, this results implied that the self-aggregated oligomers of 1 were dissociated due to the coordination between 1 and Ag+. In contrast, upon addition of Cu2+, nanospheres changed into nanofibers (Fig. 6c). With the introduction of Zn2+, nanostructures with belt-like morphology were formed (Fig. 6d). The different morphologies obtained for the self-aggregation of the molecule 1 with metal ions reflected the differences of coordination modes. Though the reasons for this unusual morphology changes were not perfectly understood, it could be assumed that the metal ions played an important role. These self-aggregations are currently being further investigated to explain the role of the metal ions in the self-aggregations process and to elucidate the exact mode of self-aggregation.
 | ||
| Fig. 6 TEM images of (a) 1, (b) 1 + Ag+, (c) 1 + Cu2+ and (d) 1 + Zn2+. | ||
Conclusions
In conclusion, we designed and synthesized three pyrimidine-containing claw-shaped unusual hexaarylbenzene derivatives (HABs) by cobalt-catalyzed cyclotrimerization of corresponding tolan derivatives. Moreover, the well-defined morphological transformation of 1 was successfully realized by addition of different metal ions. Furthermore, efforts are now in progress to investigate the full scope of the transformation to obtain a detailed mechanism and to apply this method to other coordination systems.Experimental section
General information: unless otherwise stated, all reactions were carried out under an atmosphere of argon. Commercially available chemicals were used as received. 5-Bromo-2-dodecylpyrimidine (4)24 and 3,3′-diboronatetolane (5)25 were prepared as reported in the literature. 1H and 13C NMR spectra were recorded in CDCl3 on Bruker Model Avance DMX 400 (400 MHz) at ambient temperature. Chemical shifts in NMR were reported in ppm (δ), relative to the internal standard of tetramethylsilane (TMS). Matrix-assisted laser desorption-ionization time-of-flight mass spectrometry (MALDI-TOF mass) was recorded on 4800 Module Diagnostics from ABSCIEX. Electronic absorption spectra were recorded on a PERSEE model TU-1901 spectrophotometer using a quartz cell of 1 cm path length. Fluorescence spectra were recorded on a HORIBA model Fluoromax-4 spectrophotometer. All new compounds were characterized by 1H NMR, 13C NMR, and MALDI-TOF mass.Synthesis of 6
Under an argon atmosphere, to a 100 mL flask were added 4 (689 mg, 2.11 mmol), 5 (230 mg, 0.53 mmol), Pd(PPh3)4 (30 mg, 0.026 mmol), 2 M K2CO3 aqueous solution (6 mL, 7.95 mmol) and toluene (15 mL), respectively. The mixture was stirred at 90 °C for 12 h. After being cooled to room temperature, the reaction mixture was concentrated under reduced pressure. The residue was washed with water and extracted with CH2Cl2. The organic layer was dried over anhydrous MgSO4, filtered, and concentrated to afford the crude product, which was purified by column chromatography (SiO2, EA/CH2Cl2 = 1:40) to allow isolation of 6 as a white solid in 75% yield. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.91 (s, 4H), 7.76 (s, 2H), 7.63 (d, J = 7.2 Hz, 2H), 7.58–7.49 (m, 4H), 3.03 (t, J = 7.6 Hz, 4H), 1.95–1.80 (m, 4H), 1.26–1.24 (m, 36H), 0.88 (t, J = 6.6 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ (ppm): 170.59, 154.93, 134.88, 131.81, 130.51, 129.94, 129.52, 126.95, 124.18, 89.59, 39.11, 31.92, 30.90, 29.66, 29.64, 29.54, 29.46, 29.42, 29.35, 28.78, 22.68, 14.10. MALDI-TOF mass: calcd for C46H62N4 [M]+: m/z = 671.01, found: 671.53.
General procedure of cyclotrimerization
A suspension of tolan derivatives (1 eq.) in dioxane was degassed several times with argon, then Co2(CO)8 (0.15 eq.) was added. The resulting mixture was refluxed for 12 hours until starting materials disappeared, as detected by TLC. While cooling to room temperature, the mixture was poured into water and extracted with CH2Cl2 (3 × 20 mL). The organic extracts was washed with brine and dried over MgSO4, and the solvent was removed under reduced pressure.Synthesis of 1
Co2(CO)8 (15.5 mg, 0.045 mmol) was added to a suspension of 6 (200 mg, 0.30 mmol) in dioxane (30 mL). The resulting mixture was refluxed for 12 hours and treated as described in the general procedure. Purification by column chromatography on silica gel with acetone–CH2Cl2 (1:5) gave 180 mg (90%) 1 as a white solid. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.34 (t, J = 12.3 Hz, 12H), 7.25–6.93 (m, 24H), 2.92 (t, J = 7.7 Hz, 12H), 1.89–1.67 (m, 12H), 1.41–1.20 (m, 108H), 0.86 (t, J = 6.7 Hz, 18H). 13C NMR (101 MHz, CDCl3) δ (ppm): 170.32, 154.71, 154.67, 141.05, 140.32, 133.88, 133.71, 131.73, 130.68, 130.11, 128.42, 124.76, 39.19, 31.91, 30.91, 29.67, 29.65, 29.62, 29.56, 29.53, 29.50, 29.47, 29.45, 29.35, 28.89, 28.81, 22.68, 14.11. MALDI-TOF mass: calcd for C138H186N12 [M]+: m/z = 2013.09, found: 2013.62.
Synthesis of 9
The mixture of 4 (200 mg, 0.578 mmol), 8 (145 mg, 0.53 mmol), Pd(PPh3)4 (50 mg, 0.0525 mmol), 2 M K2CO3 aqueous solution (2.62 mL, 5.3 mmol) and toluene (15 mL) was degassed by three “freeze–pump–thaw” cycles, and the resulting mixture was heated at 90 °C under an argon atmosphere for 12 h. After being cooled to room temperature, the solvent was then removed under reduced pressure. The residue was washed with water and extracted with CH2Cl2. The organic layer was dried over anhydrous MgSO4, filtered, and concentrated to afford the crude product, which was purified by column chromatography (SiO2, CH2Cl2/PE = 1:2) to allow isolation of 9 as a white solid in 77% yield. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.86 (s, 2H), 7.66 (s, 1H), 7.60 (d, J = 7.0 Hz, 1H), 7.56–7.46 (m, 2H), 3.01 (t, J = 7.7 Hz, 2H), 1.90–1.82 (m, 2H), 1.48–1.18 (m, 18H), 0.88 (t, J = 6.6 Hz, 3H), 0.32 (s, 9H). 13C NMR (101 MHz, CDCl3) δ (ppm): 170.22, 155.06, 142.10, 133.97, 133.57, 131.72, 131.57, 128.62, 127.30, 39.21, 31.92, 29.67, 29.64, 29.55, 29.48, 29.44, 29.35, 28.80, 22.68, 14.10. MALDI-TOF mass: calcd for C25H40N2Si [M]+: m/z = 396.68, found: 397.23.
Synthesis of 10
A solution of ICl (1.2 mL, 1.0 M in CH2Cl2, 1.2 mmol) was added dropwise to a solution of 9 (160 mg, 0.40 mmol) in CH2Cl2 (10 mL) at 0 °C. The solution was stirred overnight at room temperature under darkness, saturated sodium thiosulfate was added until the reaction mixture turned to colorless, the organic layer separated, the aqueous layer extracted with CH2Cl2, and the combined organic layer dried over MgSO4 and evaporated to dryness. The residue was purified by column chromatography (SiO2, EA/PE = 1/10) to give 10 as a white solid in 88% yield. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.83 (s, 2H), 7.91 (s, 1H), 7.77 (d, J = 7.8 Hz, 1H), 7.52 (d, J = 7.6 Hz, 1H), 7.22 (d, J = 7.8 Hz, 1H), 3.03 (t, J = 7.9 Hz, 2H), 1.91–1.81 (m, 2H), 1.38–1.20 (m, 18H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ (ppm): 170.91, 154.90, 137.59, 136.81, 135.75, 130.90, 129.82, 126.10, 95.11, 39.22, 31.92, 29.66, 29.63, 29.54, 29.45, 29.41, 29.35, 28.76, 22.68, 14.10. MALDI-TOF mass: calcd for C22H31N2I [M]+: m/z = 450.40, found: 447.28.Synthesis of 11
The mixture of 1-bromo-4-dodecylbenzene (2.00 g, 6.20 mmol), bis(pinacolato)diboron (3.5 g, 12.4 mmol), PdCl2(dppf) (220 mg, 0.30 mmol), KOAc (1.75 g, 17.9 mmol) and DMF (50 mL) was degassed by three “freeze–pump–thaw” cycles, and the resulting mixture was heated at 90 °C under an argon atmosphere for 12 h. After being cooled to room temperature, the solvent was then removed under reduced pressure. The residue was washed with water and extracted with CH2Cl2. The organic layer was dried over anhydrous MgSO4, filtered, and concentrated to afford the crude product, which was purified by column chromatography (SiO2, PE/CH2Cl2 = 10:1) to give 11 as colorless oil in 78% yield. 1H NMR (400 MHz, CDCl3) δ (ppm): 7.75 (d, J = 7.6 Hz, 2H), 7.22 (d, J = 7.6 Hz, 2H), 2.63 (t, J = 7.7 Hz, 2H), 1.67–1.57 (m, 2H), 1.36 (s, 12H), 1.29–1.25 (m, 18H), 0.90 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ (ppm): 146.42, 134.80, 127.89, 83.59, 36.20, 31.93, 31.35, 29.67, 29.66, 29.59, 29.51, 29.36, 29.32, 25.03, 24.86, 22.69, 14.12. MALDI-TOF mass: calcd for C24H41BO2 [M]+: m/z = 372.39, found: 374.25.
Synthesis of 12
A mixture of 11 (1 g, 2.69 mmol), 1-bromo-3-iodobenzene (1.14 g, 4.03 mmol.), Pd(PPh3)4 (150 mg, 0.14 mmol), 2 M K2CO3 aqueous solution (13.5 mL, 27 mmol) and THF (40 mL) was degassed by three “freeze–pump–thaw” cycles, and the resulting mixture was heated at 50 °C under an argon atmosphere for 12 h. After being cooled to room temperature, the solvent was then removed under reduced pressure. The residue was washed with water and extracted with CH2Cl2. The organic layer was dried over anhydrous MgSO4, filtered, and concentrated to afford the crude product, which was purified by column chromatography (SiO2, PE) to give 12 as colorless liquid in 81% yield. 1H NMR (400 MHz, CDCl3) δ (ppm): 7.75 (s, 1H), 7.55–7.44 (m, 4H), 7.32–7.27 (m, 3H), 2.66 (t, J = 7.8 Hz, 2H), 1.70–1.62 (m, 2H), 1.36–1.24 (m, 18H), 0.91 (t, J = 6.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ (ppm): 143.37, 142.85, 137.02, 130.19, 130.04, 129.86, 128.96, 126.94, 125.56, 122.87, 35.64, 31.96, 31.47, 29.71, 29.70, 29.68, 29.63, 29.55, 29.39, 29.37, 22.72, 14.14. MALDI-TOF mass: calcd for C24H33Br [M]+: m/z = 401.42, found: 400.33.Synthesis of 15
The mixture of 12 (900 mg, 2.25 mmol), TMSA (460 mg, 4.5 mmol), Pd(PPh3)2Cl2 (63 mg, 5% mmol), CuI (21 mg, 5% mmol) and Et3N (40 mL) was degassed by three “freeze–pump–thaw” cycles, and the resulting mixture was heated at 80 °C under an argon atmosphere for 12 h. After being cooled to room temperature, the solvent was then removed under reduced pressure. The residue was washed with water and extracted with CH2Cl2. The organic layer was dried over anhydrous MgSO4, filtered, and concentrated to afford the crude product, which was purified by column chromatography (SiO2, PE) to give 13 as colorless liquid.The solution of tetrabutylammonium fluoride (TBAF) (940 mg, 2.25 mmol) in THF (2 mL) was added into a solution of 13 (400 mg) in THF (10 mL). The resulting mixture was stirred under an argon atmosphere for 5 min and poured into saturated ammonium chloride solution. The mixture was extracted twice with ethyl acetate. The combined extracts were washed with water and dried over MgSO4. After the solvent was removed, 312 mg product was obtained, which was used in the next step without further purification.
The mixture of 10 (312 mg, 0.693 mmol), 14 (242 mg, 0.693 mmol), Pd(PPh3)2Cl2 (25 mg, 0.037 mmol), CuI (7 mg, 0.039 mmol) and Et3N (40 mL) was degassed by three “freeze–pump–thaw” cycles, and the resulting mixture was heated at 70 °C under an argon atmosphere for 12 h. After being cooled to room temperature, the solvent was then removed under reduced pressure. The residue was washed with water and extracted with CH2Cl2. The organic layer was dried over anhydrous MgSO4, filtered, and concentrated to afford the crude product, which was purified by column chromatography (SiO2, EA/PE = 1/8) to give 15 as champagne solid in 70% yield. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.90 (s, 2H), 7.77 (d, J = 14.6 Hz, 2H), 7.63–7.40 (m, 9H), 7.28 (s, 1H), 3.03 (t, J = 7.5 Hz, 2H), 2.65 (t, J = 7.6 Hz, 2H), 1.92–1.85 (m, 2H), 1.70–1.60 (m, 2H), 1.48–1.22 (m, 36H), 0.88 (t, J = 6.3 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ (ppm): 170.64, 154.95, 142.61, 141.48, 137.52, 134.88, 131.72, 130.52, 130.23, 130.11, 129.89, 129.42, 128.91, 128.81, 127.26, 126.92, 126.63, 124.59, 123.23, 90.54, 88.57, 39.21, 35.63, 31.92, 31.46, 29.67, 29.64, 29.60, 29.55, 29.52, 29.47, 29.44, 29.36, 28.79, 22.69, 14.10. MALDI-TOF mass: calcd for C48H64N2 [M]+: m/z = 669.04, found: 669.62.
Synthesis of 2 and 3
Co2(CO)8 (15.5 mg, 0.045 mmol) was added to a suspension of 15 (300 mg, 0.30 mmol) in dioxane (30 mL). The resulting mixture was refluxed for 12 hours and treated as described in the general procedure. The mixture could hardly be separated by column chromatography because of their similar polarities. The ratio of EA/DCM/PE = 1:10:10 was adapted to separate the isomers in thin-layer chromatography. Two isomers 2 and 3 could be separated by repeatedly preparative TLC in this solvent system to get the final product 2 in 27% yields as white solid and 3 in 56% yields as a white solid.
Compound 2: 1H NMR (400 MHz, CDCl3) δ (ppm): 8.39 (s, 6H), 7.21–6.82 (m, 36H), 2.98–2.88 (m, 6H), 2.60–2.50 (m, 6H), 1.86–1.72 (m, 6H), 1.62–1.52 (m, 6H), 1.36–1.24 (m, 108H), 0.86 (t, J = 6.2 Hz, 18H). MALDI-TOF mass: calcd for C144H192N6 [M]+: m/z = 2007.11, found: 2007.92.
Compound 3: 1H NMR (400 MHz, CDCl3) δ (ppm): 8.39 (s, 6H), 7.19–6.87 (m, 36H), 2.98–2.92 (m, 6H), 2.58–2.50 (m, 6H), 1.85–1.75 (m, 6H), 1.62–1.52 (m, 6H), 1.32–1.18 (m, 108H), 0.86 (t, J = 6.3 Hz, 18H). MALDI-TOF mass: calcd for C144H192N6 [M]+: m/z = 2007.11, found: 2007.83.
Acknowledgements
We are grateful for financial support provided by the National Science Foundation of China (21172035, 21302018); Research Fund for the Doctoral Program of Higher Education of China (20120075120017, 20120075110001); The Fundamental Research Funds for the Central Universities (13D110529, 13D111313).Notes and references
- A. Narita, X. Wang, X. Feng and K. Müllen, New Advances in Nanographene Chemistry, Chem. Soc. Rev., 2015, 44, 6616 RSC.
- (a) K. Sakajiri, T. Sugisaki and K. Moriya, Stable Supramolecular Helical Structure of C6-symmetric Hydrogen-bonded Hexakis(Phenylethynyl)Benzene Derivatives with Amino Acid Pendant Groups and Their Uniqe Fluorescence Properties, Chem. Commun., 2008, 3447 RSC; (b) S. Prasanthkumar, W. Zhang, W. Jin, T. Fukushima and T. Aida, Selective Synthesis of Single- and Multi-Walled Supramolecular Nanotubes by Using Solvophobic/Solvophilic Controls: Stepwise Radial Growth via “Coil-on-Tube” Intermediates, Angew. Chem., Int. Ed., 2015, 54, 11168 CrossRef CAS PubMed.
- (a) D. Rausch and C. Lambert, Synthesis and Spectroscopic Properties of a Hexapyrenylbenzene Derivative, Org. Lett., 2006, 8, 5037 CrossRef CAS PubMed; (b) G. Bottari and T. Torres, Synthesis and Characterization of a Benzene-Centered, Phthalocyanine Hexamer, Chem. Commun., 2004, 2668 RSC.
- (a) M. D. Watson, A. Fechtenkötter and K. Müllen, Big Is Beautiful-“Aromaticity” Revisited from the Viewpoint of Macromolecular and Supramolecular Benzene Chemistry, Chem. Rev., 2001, 101, 1267 CrossRef CAS PubMed; (b) S. Suzuki, Y. Segawa, K. Itami and J. Yamaguchi, Synthesis and Characterization of Hexaarylbenzenes with Five or Six Different Substituents Enabled by Programmed Synthesis, Nat. Chem., 2015, 7, 227 CrossRef CAS PubMed.
- H. S. Cho, H. Rhee, J. K. Song, C. Min, M. Takase, N. Aratani, S. Cho, A. Osuka, T. Joo and D. Kim, Excitation Energy Transport Processes of Porphyrin Monomer, Dimer, Cyclic Trimer, and Hexamer Probed by Ultrafast Fluorescence Anisotropy Decay, J. Am. Chem. Soc., 2003, 125, 5849 CrossRef CAS PubMed.
- (a) S. Ito, H. Inabe, N. Morita, K. Ohta, T. Kitamura and K. Imafuku, Synthesis of Poly(6-azulenylethynyl)benzene Derivatives as a Multielectron Redox System with Liquid Crystalline Behavior, J. Am. Chem. Soc., 2003, 125, 1669 CrossRef CAS PubMed; (b) S. Ito, M. Ando, A. Nomura, N. Morita, C. Kabuto, H. Mukai, K. Ohta, J. Kawakami, A. Yoshizawa and A. Tajiri, Synthesis and Properties of Hexakis(6-octyl-2-azulenyl)benzene as a Multielectron Redox System with Liquid Crystalline Behavior, J. Org. Chem., 2005, 70, 3939 CrossRef CAS PubMed; (c) V. J. Chebny, D. Dhar, S. V. Lindeman and R. Rathore, Simultaneous Ejection of Six Electrons at a Constant Potential by Hexakis(4-ferrocenylphenyl)benzene, Org. Lett., 2006, 8, 5041 CrossRef CAS PubMed; (d) R. Rathore, C. L. Burns and M. I. Deselnicu, Multiple-Electron Transfer in a Single Step. Design and Synthesis of Highly Charged Cation-Radical Salts, Org. Lett., 2001, 3, 2887 CrossRef CAS PubMed; (e) C. Lambert, Hexaarylbenzenes-Prospects for Toroidal Delocalization of Charge and Energy, Angew. Chem., Int. Ed., 2005, 44, 7337 CrossRef CAS PubMed.
- (a) K. Kobayashi, A. Sato, S. Sakamoto and K. Yamaguchi, Solvent-induced Polymorphism of Three-dimensional Hydrogen-bonded Networks of Hexakis(4-carbamoylphenyl)benzene, J. Am. Chem. Soc., 2003, 125, 3035 CrossRef PubMed; (b) K. E. Maly, E. Gagnon, T. Maris and J. D. Wuest, Engineering Hydrogen-bonded Molecular Crystals Built from Derivatives of Hexaphenylbenzene and Related Compounds, J. Am. Chem. Soc., 2007, 129, 4306 CrossRef PubMed.
- (a) B. Schmaltz, T. Weil and K. Müllen, Polyphenylene-based Materials: Control of the Electronic Function by Molecular and Supramolecular Complexity, Adv. Mater., 2009, 21, 1067 CrossRef CAS; (b) A. C. Grimsdale and K. Müllen, Bridged Polyphenylenes from Polyfluorenes to Ladder Polymers, Adv. Polym. Sci., 2008, 212, 1 Search PubMed; (c) A. C. Grimsdale and K. Müllen, Oligomers and Polymers Based on Bridged Phenylenes as Electronic Materials, Macromol. Rapid Commun., 2007, 28, 1676 CrossRef CAS; (d) Y. Geng, A. Fechtenkötter and K. Müllen, Star-like Substituted Hexaarylbenzenes: Synthesis and Mesomorphic Properties, J. Mater. Chem., 2001, 11, 1634 RSC.
- (a) V. J. Chebny, R. Shukla and R. Rathore, Toroidal Hopping of a Single Hole Through the Circularly-Arrayed Naphthyl Groups in Hexanaphthylbenzene Cation Radical, J. Phys. Chem. A, 2006, 110, 13003 CrossRef CAS PubMed; (b) S. V. Rosokha, I. S. Neretin, D. Sun and J. K. Kochi, Very Fast Electron Migrations Within p-Doped Aromatic Cofacial Arrays Leading to Three-Dimensional (Toroidal) π-Delocalization, J. Am. Chem. Soc., 2006, 128, 9394 CrossRef CAS PubMed; (c) V. Mamane, A. Gref, F. Lefloch and O. Riant, [2 + 2 + 2] Cyclotrimerisation of Bisaryl Acetylene Bearing Ferrocenyl Units with Planar Chirality: Synthesis of Enantiopure Conjugated Polyferrocene Complexes, J. Organomet. Chem., 2001, 637–639, 84 CrossRef CAS.
- S. S. Gunathilake, H. D. Magurudeniya, P. Huang, H. Nguyen, E. A. Rainbolt, M. C. Stefan and M. C. Biewer, Synthesis and Characterization of Novel Semiconducting Polymers Containing Pyrimidine, Polym. Chem., 2013, 4, 5216 RSC.
- V. F. Petrov, Pyrimidine as a Structural Fragment in Calamitic Liquid Crystals, Mol. Cryst. Liq. Cryst., 2006, 457, 121 CrossRef CAS.
- (a) C. Wang, G. Y. Jung, A. S. Batsanov, M. R. Bryce and M. C. Petty, New Electron-transporting Materials for Light Emitting Diodes: 1,3,4-Oxadiazole-pyridine and 1,3,4-Oxadiazole-pyrimidine Hybrids, J. Mater. Chem., 2002, 12, 173 RSC; (b) G. H. Kim, R. Lampande, J. H. Kong, J. M. Lee, J. H. Kwon, J. K. Lee and J. H. Park, New Bipolar Host Materials for High Performance of Phosphorescent Green Organic Light-Emitting Diodes, RSC Adv., 2015, 5, 31282 RSC.
- (a) C. Tang, Q. Zhang, D. Li, J. Zhang, P. Shi, S. Li, J. Wu and Y. Tian, Synthesis, Crystal Structures, Two-photon Absorption and Biological Imaging Application of Two Novel Bent-shaped Pyrimidine Derivatives, Dyes Pigm., 2013, 99, 20 CrossRef CAS; (b) K. K. Upadhyay and A. Kumar, Pyrimidine Based Highly Sensitive Fluorescent Receptor for Al3+ Showing Dual Signalling Mechanism, Org. Biomol. Chem., 2010, 8, 4892 RSC.
- C. Hadad, S. Achelle, I. López-Solera, J. C. García-Martínez and J. Rodríguez-López, Metal Cation Complexation Studies of 4-Arylvinyl-2,6-di(Pyridin-2-yl)Pyrimidines: Effect on the Optical Properties, Dyes Pigm., 2013, 97, 230 CrossRef CAS.
- (a) S. M. Draper, D. J. Gregg and R. Madathil, Heterosuperbenzenes: A New Family of Nitrogen-Functionalized, Graphitic Molecules, J. Am. Chem. Soc., 2002, 124, 3486 CrossRef CAS PubMed; (b) D. J. Gregg, C. M. A. Ollagnier, C. M. Fitchett and S. M. Draper, Structurally Characterized Hetero-Oligopolyphenylenes: Synthetic Advances Toward Next-Generation Heterosuperbenzenes, Chem.–Eur. J., 2006, 12, 3043 CrossRef CAS PubMed.
- (a) S. M. Draper, D. J. Gregg, E. R. Schofield, W. R. Browne, M. Duati, J. G. Vos and P. Passaniti, Complexed Nitrogen Heterosuperbenzene: the Coordinating Properties of a Remarkable Ligand, J. Am. Chem. Soc., 2004, 126, 8694 CrossRef CAS PubMed; (b) C. M. A. Ollagnier, S. D. Perera, C. M. Fitchett and S. M. Draper, Rhodium and Palladium Complexes of a Pyridyl-centred Polyphenylene Derivative, Dalton Trans., 2008, 2, 283 RSC; (c) K. Mitsudo, J. Harada, Y. Tanaka, H. Mandai, C. Nishioka, H. Tanaka, A. Wakamiya, Y. Murata and S. Suga, Synthesis of Hexa(Furan-2-yl)Benzenes and Their π-Extended Derivatives, J. Org. Chem., 2013, 78, 2763 CrossRef CAS PubMed.
- N. E. Schore, Transition Metal-mediated Cycloaddition Reactions of Alkynes in Organic Synthesis, Chem. Rev., 1988, 88, 1081 CrossRef CAS.
- M. Steeger and C. Lambert, Charge-Transfer Interactions in Tris-Donor-Tris-Acceptor Hexaarylbenzene Redox Chromophores, Chem.–Eur. J., 2012, 18, 11937 CrossRef PubMed.
- P. Kissel, S. Breitler, V. Reinmüller, P. Lanz, L. Federer, A. D. Schlüter and J. Sakamoto, An Easy and Multigram-Scale Synthesis of Versatile AA- and AB-type m-Terphenylenes as Building Blocks for Kinked Polyphenylenes, Eur. J. Org. Chem., 2009, 18, 2953 CrossRef.
- (a) A. Steffen, R. M. Ward, W. D. Jones and T. B. Marder, Dibenzometallacyclopentadienes, Boroles and Selected Transition Metal and Main Group Heterocyclopentadienes: Synthesis, Catalytic and Optical Properties, Coord. Chem. Rev., 2010, 254, 1950 CrossRef CAS; (b) N. E. Schore, Transition-Metal-Mediated Cycloaddition Reactions of Alkynes in Organic Synthesis, Chem. Rev., 1988, 88, 1081 CrossRef CAS.
- (a) C. Lambert and G. Nöll, Optically and Thermally Induced Electron Transfer Pathways in Hexakis[4-(N,N-diarylamino)phenyl]benzene Derivatives, Chem.–Eur. J., 2002, 8, 3467 CrossRef CAS PubMed; (b) C. Lambert, J. Ehbets, D. Rausch and M. Steeger, Charge-transfer Interactions in a Multichromophoric Hexaarylbenzene Containing Pyrene and Triarylamines, J. Org. Chem., 2012, 77, 6147 CrossRef CAS PubMed; (c) X. Feng, W. Pisula, T. Kudernac, D. Wu, L. Zhi, S. D. Feyter and K. Müllen, Controlled Self-Assembly of C3-Symmetric Hexa-Peri-Fexabenzocoronenes with Alternating Hydrophilic and Hydrophobic Substituents in Solution, in the Bulk, and on a Surface, J. Am. Chem. Soc., 2009, 131, 4439 CrossRef CAS PubMed.
- (a) B. Therrien, Coordination Chemistry of 2,4,6-Tri(pyridyl)-1,3,5-triazine Ligands, J. Organomet. Chem., 2011, 696, 637 CrossRef CAS; (b) T. R. Cook, Y. Zheng and P. J. Stang, Metal-organic Frameworks and Self-assembled Supramolecular Coordination Complexes: Comparing and Contrasting the Design, Synthesis, and Functionality of Metal-organic Materials, Chem. Rev., 2013, 113, 734 CrossRef CAS PubMed; (c) I. A. Bhat, D. Samanta and P. S. Mukherjee, A Pd24 Pregnant Molecular Nanoball: Self-Templated Stellation by Precise Mapping of Coordination Sites, J. Am. Chem. Soc., 2015, 137, 9497 CrossRef CAS PubMed.
- D. Bai, X. Liu and S. Wang, Charge-transfer Emission Involving Three-coordinate Organoboron: V-shape versus U-shape and Impact of the Spacer on Dual Emission and Fluorescent Sensing, Chem.–Eur. J., 2007, 13, 5713 CrossRef CAS PubMed.
- Y. Zhang, Y. Wu, D. Zhang and W. Jin, Selective Synthesis of Novel 5-Bromopyrimidine Derivatives, Chin. J. Synth. Chem., 2011, 19, 662 Search PubMed.
- M. Takase, A. Nakajima and T. Takeuchi, Synthesis of an Extended Hexagonal Molecule as a Highly Symmetrical Ligand, Tetrahedron Lett., 2005, 46, 1739 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra18422c |
| This journal is © The Royal Society of Chemistry 2015 |