
A high-throughput synthesis of 1,2,4-oxadiazole and 1,2,4-triazole libraries in a continuous flow reactor†
Abstract
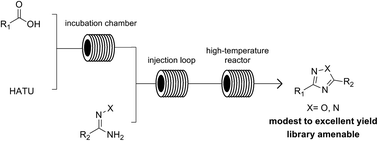
We report herein a high-throughput methodology for the synthesis of 1,2,4-oxadiazole and 1,2,4-triazole small-molecule libraries using an integrated synthesis and purification platform. The heterocyclization relies first on a low-temperature peptide coupling of a diverse set of carboxylic acids and hydroxyamidines, hydrazonamides, or pyridyl hydrazides followed by a high-temperature cyclization to yield the respective heterocycles in a continuous flow process. The fully integrated synthesis and purification platform enables the rapid generation of chemical libraries, decreasing the drug discovery cycle time.
 Please wait while we load your content...
Please wait while we load your content...

