
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Saturation transfer difference NMR to study substrate and product binding to human UDP-xylose synthase (hUXS1A) during catalytic event
Claudia
Puchner
a,
Thomas
Eixelsberger
b,
Bernd
Nidetzky
b and
Lothar
Brecker
*a
aUniversity of Vienna, Institute of Organic Chemistry, Währingerstrasse 38, A-1090 Vienna, Austria. E-mail: lothar.brecker@univie.ac.at; Fax: +43 1 4277 9521; Tel: +43 1 4277 52131
bGraz University of Technology, Institute of Biotechnology and Biochemical Engineering, Petersgasse 12/1, A-8010 Graz, Austria
First published on 29th September 2015
Abstract
The human form of UDP-xylose synthase (hUXS1A) is studied with respect to its substrate and co-enzyme binding in binary and ternary complexes using saturation transfer difference (STD) NMR and in situ NMR. Obtained binding pattern results are correlated to the recently solved crystal structure of hUXS1A and docking studies of UDP-GlcUA, providing a better understanding of substrate specificity of this enzyme and may give useful information in mutant designing. In unproductive binary complexes UDP-saccharide aglycone moieties show strong STD effects with the protein. In contrast, pyranoside rings (Glc, GlcUA, Gal) indicate less interaction with the hUXS1A active site, which enables the required ring distortion of the pyranoside ring in UDP-GlcUA. In productive ternary complexes UDP-GlcUA possesses reasonable binding, while produced UDP-Xyl shows smaller STD responses and does not efficiently compete with the substrate for binding at the active site. STD NMR derived binding studies of NAD+ demonstrate tight interaction between co-factor and hUXS1A. Higher magnetization of NAD+ in the presence of enzymatic product is observed and suggests increased contact with groups on the protein. Furthermore, binding studies of substrate analogues having the same stereochemistry as the investigated UDP-saccharides and a small aglycone residue indicate a different mode of action, not guided by the anchor groups.
1. Introduction
Uridine diphosphate D-glucuronic acid (UDP-GlcUA) is an important carbohydrate being of common interest because of its position in several detoxification processes and biosynthetic pathways.1,2 It functions as a donor-substrate for various UDP-glucuronosyl transferases and supports the incorporation of D-glucuronosyl residues into nascent glycosaminoglycans, like hyaluronan.3 UDP-GlcUA is also involved in O-glucuronidation of small molecules in xenobiotic metabolism.2 Further, it acts as substrate in the formation of several carbohydrates in different organisms. Varying metabolic pathways depend on different enzymatic catalysis as shown in Scheme 1.4–8 The currently most intensively studied pathway is the mammalian UDP-xylose synthase (UXS) catalyzed formation of UDP-xylose (UDP-Xyl). This product is of importance in several living organisms as it acts as a precursor for several glycan structures in mammals, plants, fungi as well as in bacteria.4,9 It is, however, generated by various alternative routes in different species. In mammals, formation of UDP-Xyl stimulates the synthesis of glycosaminoglycans on the protein core of extracellular matrix proteoglycans. Due to its necessary role as receptors, proteoglycans are involved in cell signaling pathways including tissue development. A lack of UXS hence leads to modifications in extracellular matrix, causing changes in morphogenesis of different tissues, which may lead to e.g. tumor growth and progression.10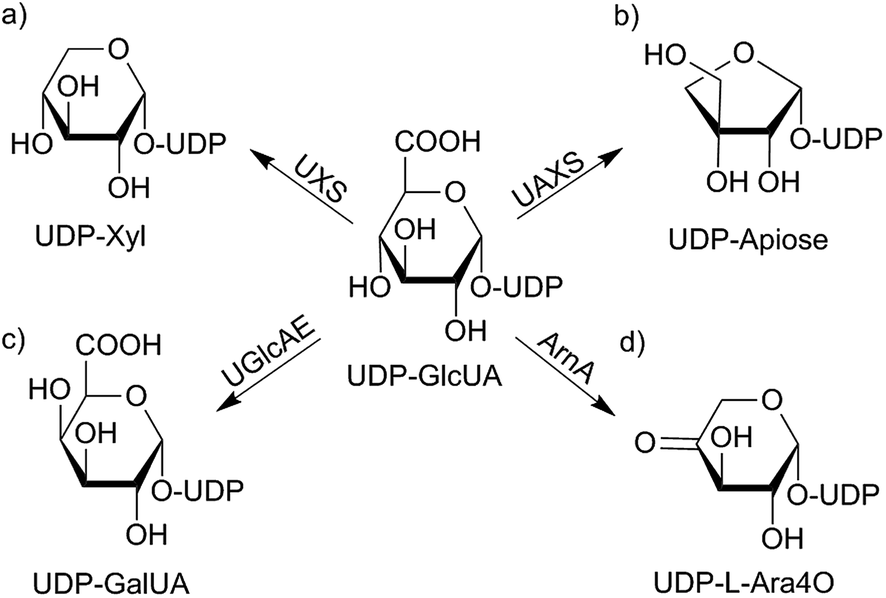 | ||
| Scheme 1 UDP-GlcUA metabolism depending on different enzyme sources from varying organisms: in mammals, UDP-GlcUA gets converted to UDP-Xyl by NAD+ dependent glucuronic acid decarboxylase (hUXS1A) (a). In plants substrate is turned into the C-3-branched sugar UDP-apiose (b) as well as to UDP-galacturonic acid (UDP-GalUA) (c). Further, a polymyxin-resistant mutant of Escherichia coli is able to transform UDP-GlcUA into UDP-L-4-keto-arabinose (UDP-L-Ara4O) (d).4–9 | ||
The human form of UDP-xylose synthase (hUXS1A) exclusively converts UDP-GlcUA to UDP-Xyl.11 Its biological active form is homodimeric and belongs to the short-chain dehydrogenase/reductase (SDR) superfamily.12 The active site of hUXS1A includes 6 different amino acid residues of Thr118, Tyr147 and Lys151 which are typical for members of the SDR-family. Further, Ser119, Glu120 and Arg277 are characteristic for the UXS enzyme group.10 Enzymatic conversion of UDP-GlcUA to UDP-Xyl contains mainly three chemical steps. First of all, NAD+ dependent oxidation at C4 of the pyranoside moiety, followed by subsequent decarboxylation yielding intermediate UDP-4-keto-xylose (UDP-Xyl-4O).13–15 Then, carbonyl function at C4 gets stereoselectively reduced to the (R)-alcohol by oxidizing intermediately formed NADH to NAD+. Finally, UDP-Xyl is received as product.10,13
Previous work of Eixelsberger et al.10 investigated the hUXS1A catalyzed mechanism, using energy-minimized docking of natural substrate UDP-GlcUA and MD simulations of ternary complex of hUXS1A·UDP-GlcUA·NAD+. Their results indicate a sugar ring distortion of low-energy 4C1 chair to BO,3 boat conformation which facilitate catalysis. Due to ring distortion, oxidation at C4 is relieved by arrangement side chain of Tyr147 with C4 hydroxyl group of UDP-GlcUA. Simultaneously, carboxylate group at C5 is brought into a nearly axial position forming hydrogen bonds to Thr118 and Ser119, promoting subsequent decarboxylation of obtained UDP-4-keto-GlcUA. Resulting 4-keto-intermediate is then stabilized as enolate in 2H1 half-chair conformation by protonated form of Tyr147. Finally, enolate is protonated si-facial at C5, employing water coordination by Glu120. Last, reduction of keto-function at C4 is achieved by NADH and supporting Tyr147 as catalytic proton donor to receive UDP-Xyl in 4C1 chair conformation.12,16
Based on its essential role in carbohydrate mechanisms, further investigation of hUXS1A transformation is of importance to gain additional knowledge of this type of enzymatic conversion. We now study binding pattern of different UDP-saccharides and corresponding glycosides to hUXS1A in binding only and productive mode, including influence of co-bound NAD+/NADH. In situ NMR, saturation transfer difference NMR (STD NMR) and combined in situ STD NMR are used for this purpose. The STD NMR experiment is a suitable method for analysis of protein and substrate interactions allowing to create binding epitope maps of ligands. Resulting STD responses give significant insights into binding areas of investigated substrates. Protons having close contact with the binding site of studied protein obtain more saturation transfer, thus receiving larger STD signal intensities than protons further away.16–20
Obtained binding pattern results are correlated to recently solved crystal structure of hUXS1A and docking studies of UDP-GlcUA,10 providing a better understanding of substrate specificity of this enzyme and may give useful information in designing various mutants.
2. Results and discussion
2.1. Binding pattern of UDP-sugars
STD NMR experiments with three UDP-saccharides (Fig. 1a) were recorded in absence of NAD+ to collect information about binding pattern of hUXS1A. However, small amounts of NAD+ from cell disruption and enzyme purification were present in all sample preparations, causing negligible transformation of investigated natural substrate UDP-GlcUA to UDP-Xyl during measurement of STD NMR spectra (ca. 3% transformation in 1 h). Additionally, binding of UDP-glucose (UDP-Glc) was studied due to its structural and stereo-chemical similarity to natural substrate and product. Arising of intermediate UDP-Xyl4O during conversion,10 led us to investigation of UDP-galactose (UDP-Gal) to study differences in binding behavior, which may be caused by stereo-chemical variations at C4. Resulting relative STD effects of studied UDP-glycosides are presented as epitope maps and shown in Fig. 2. Further, saturation transfer of representative protons is quantified using the STD amplification factor based on varying saccharide concentration (Fig. 3).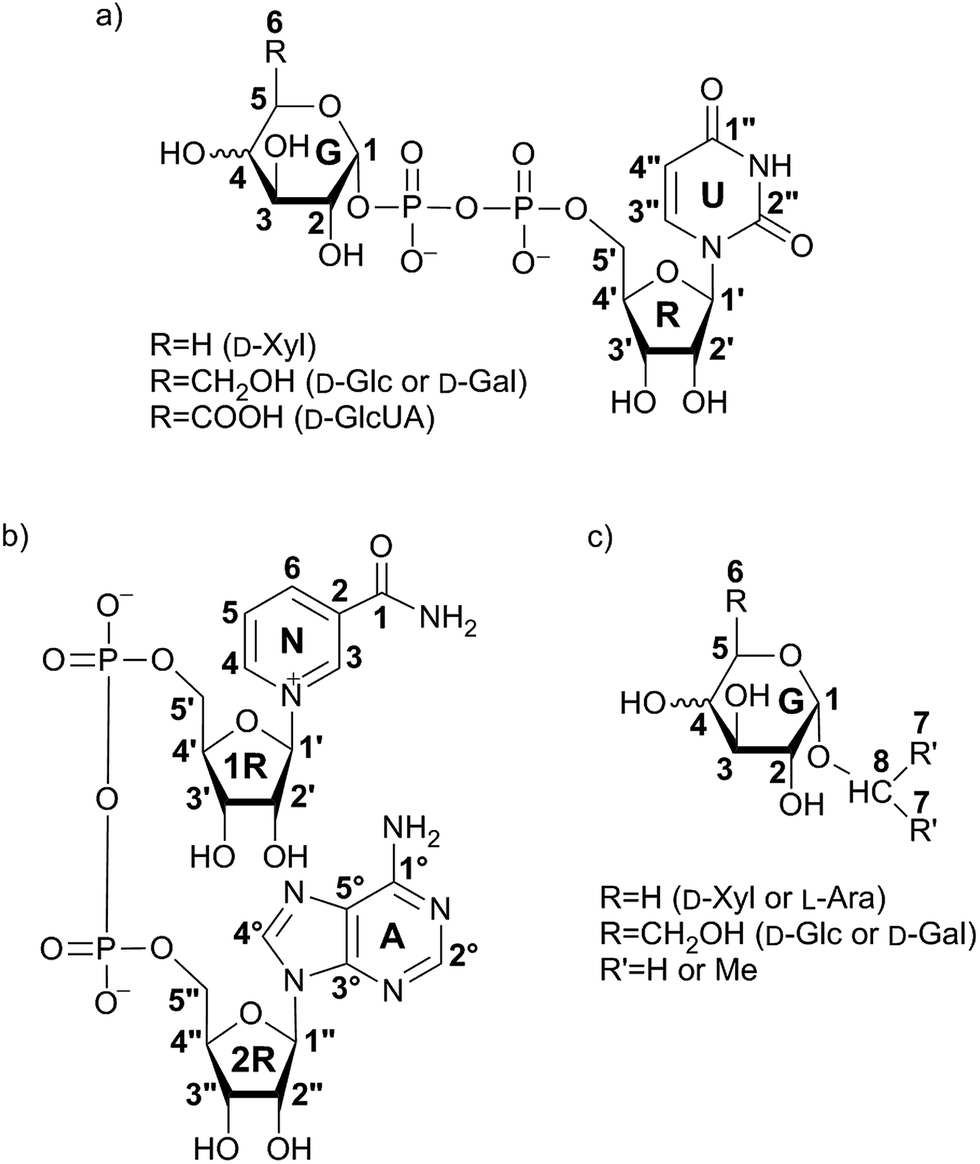 | ||
| Fig. 1 Structures of investigated UDP-saccharides (a), co-factor NAD+ (b) and synthesized α-glycosides (c). Numbering of carbon atoms does not correspond to IUPAC nomenclature numbering, but is used in Fig. 2–10 to allow comparison of STD effects for each hydrogen atom of ligands within binding to hUXS1A. | ||
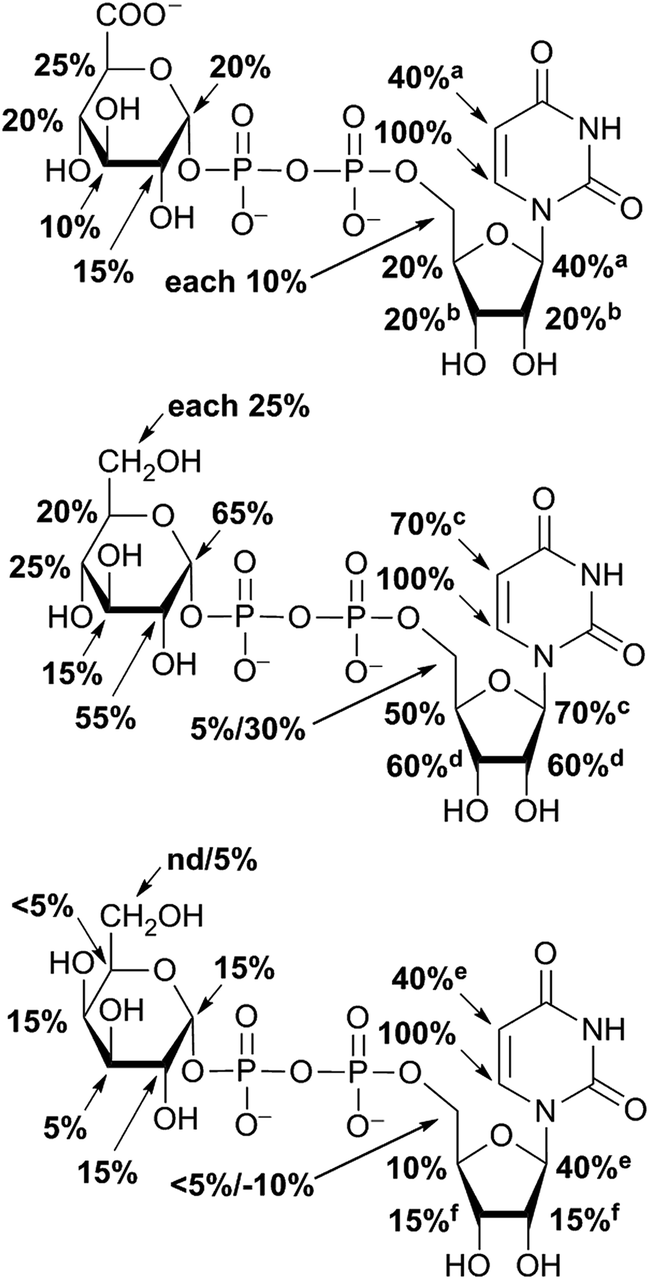 | ||
| Fig. 2 Epitope mapping of UDP-GlcUA, UDP-Glc and UDP-Gal binding to hUXS1A in absence of NAD+: aglycone moiety of UDP-saccharides possessed strong STD signals. In case of signal overlapping, STD effects are given as equal portion of combined interaction strength and indicated with superscript symbols. | ||
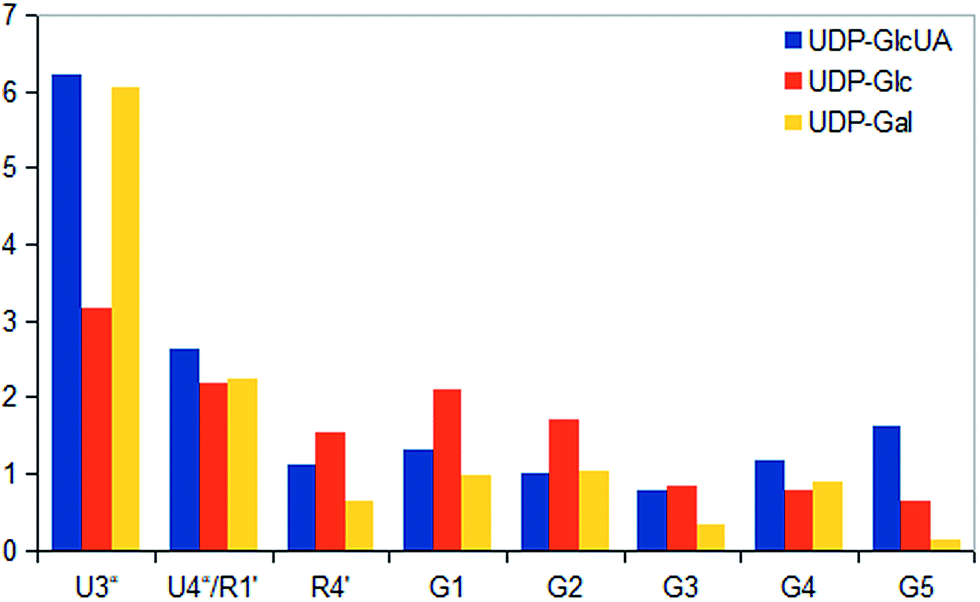 | ||
| Fig. 3 STD responses of selected protons of UDP-saccharides. Absolute values were determined as ratios between peak integrals of the STD spectra and peak integrals of the off-resonance spectra and displayed as amplification factors. | ||
Aglycone moiety of all three UDP-saccharides showed a strong response in obtained STD spectra, indicating direct contact with the binding site of hUXS1A. Especially proton U3′′ of the uracil moiety had the most intensive STD effect. This spatial closeness to the protein can be explained by π–π stacking to an aromatic protein moiety, which might be involved in the STD signal. This is in accordance with the crystal structure of hUXS1A bound with UDP, locating the uracil moiety close to aromatic residue of Tyr206.10 Additionally, anomeric proton of the ribose unit received significant saturation in all investigated UDP-sugars, suggesting this moiety also in close contact with the protein surface. In contrast, remaining ribofuranose protons had themselves smaller STD signals. Specially, low interaction between methylene group protons at R5′ and binding site were observed. This circumstance is in agreement with the X-ray structure of hUXS1A,10 given that conformation of UDP in the binding pocket leads to a partial shielding of the methylene group. Thus, having less interaction with the active site and may explain lower saturation transfer compared to other furanose protons. However, STD signals of uridine residue in investigated UDP-saccharides indicated a comparable tight contact of this moiety and hence seemed to act as an anchor, which binds the substrate and locates the pyranoside moiety close to the active site.
In general, STD responses of all three pyranoside residues of UDP-glycosides were observed and showed less interaction with binding pocket of hUXS1A compared to its uridine moiety. This circumstance is very likely due to the need for this region of substrate to be only weakly bound so that ring distortion of the pyranoside ring in UDP-GlcUA can occur during transformation. This conformational change is required for residue of Tyr147 to get hydrogen bonded with hydroxyl function at G4 for optimal positioning the reactive part of UDP-GlcUA for general base catalysis by the tyrosine.10
Nonetheless, comparison of pyranoside binding pattern of investigated UDP-saccharides demonstrated some differences in saturation transfer. In case of UDP-GlcUA protons G1, G2, G4 and G5 displayed a moderate STD response, whereas proton G3 showed decreased interaction with binding pocket of hUXS1A. These findings are supported by conducted MD simulations of UDP-GlcUA by Eixelsberger et al.10 indicating no appreciable hydrogen bond formation between G3 hydroxyl and an amino acid residue of hUXS1A, which would point to closer contact with groups on the protein.
Moreover, glucopyranoside of UDP-Glc possessed slightly modified binding compared to natural substrate. In particular, proton at G5 received decreased magnetization in contrast to G4, which let assume reduced enzymatic recognition of this area. However, whereas pyranoside proton G5 in UDP-GlcUA and UDP-Glc showed an appropriate interaction with binding pocket of hUSX1A, in contrast, change in stereochemistry at G4 led to a strong reduced STD response of G5 in UDP-Gal. At this point, it is noteworthy that proton G4 in the galactoside residue also possessed a reasonable STD effect, potentially caused by interaction with Tyr147 which is normally hydrogen bonded with C4 hydroxyl in UDP-GlcUA.10 Nonetheless, these results let suggest a suboptimal fit of this moiety to the active site or at least a partial shielding of this proton by G4 hydroxyl group and thus having less interaction with groups on the protein. Further, one proton of methylene group at position R5′′ in UDP-Gal possessed a slight negative artifact, which is presumably generated by free D2O located in a trapped position close to the ligand proton, which interferes with the ligand during saturation and spin lock event.18 Influences from slightly varying longitudinal relaxation times of ligand proton in different molecular moieties can be assumed to be small as the T1 times are in the typical range of small molecules.
2.2. STD NMR of co-factor NAD+
For further analysis of binding pattern during catalytic event, in situ NMR and in situ STD NMR investigations of hUXS1A catalyzed UDP-GlcUA transformation were performed. Apart from binding of substrate and product, NAD+ interactions with the enzyme were investigated. Therefore STD NMR of sole NAD+ bound to hUXS1A was measured. Resulting epitope mapping of sole co-factor indicated significant saturation transfer of both ribose moieties (Fig. 4). Specially, anomeric protons showed a strong response in the STD spectrum. In general, proton 2R1′′ of adenine bound furanose demonstrated more intimate contact with groups on the protein compared to proton 1R1′ of nicotinamide ribose. However, binding pattern of these riboses indicated some similarities. In both cases protons at 1R2′, 1R3′ respectively 2R3′′ possessed significant STD signals, thus having intimate interaction with the binding pocket. These findings are in agreement with X-ray structure of hUXS1A bound with NAD+,10 showing hydrogen bond formation between hydroxyl groups at 1R2′ and Tyr147, 1R3′ and Lys151 respectively 2R2′′ and Gly40 as well as 2R3′′ and Gly14. Unfortunately, STD response of proton 2R2′′ was not determinable based on change in signal intensity caused by water suppression. Nevertheless, at this point it can be assumed that this proton also is in close contact with the enzyme surface.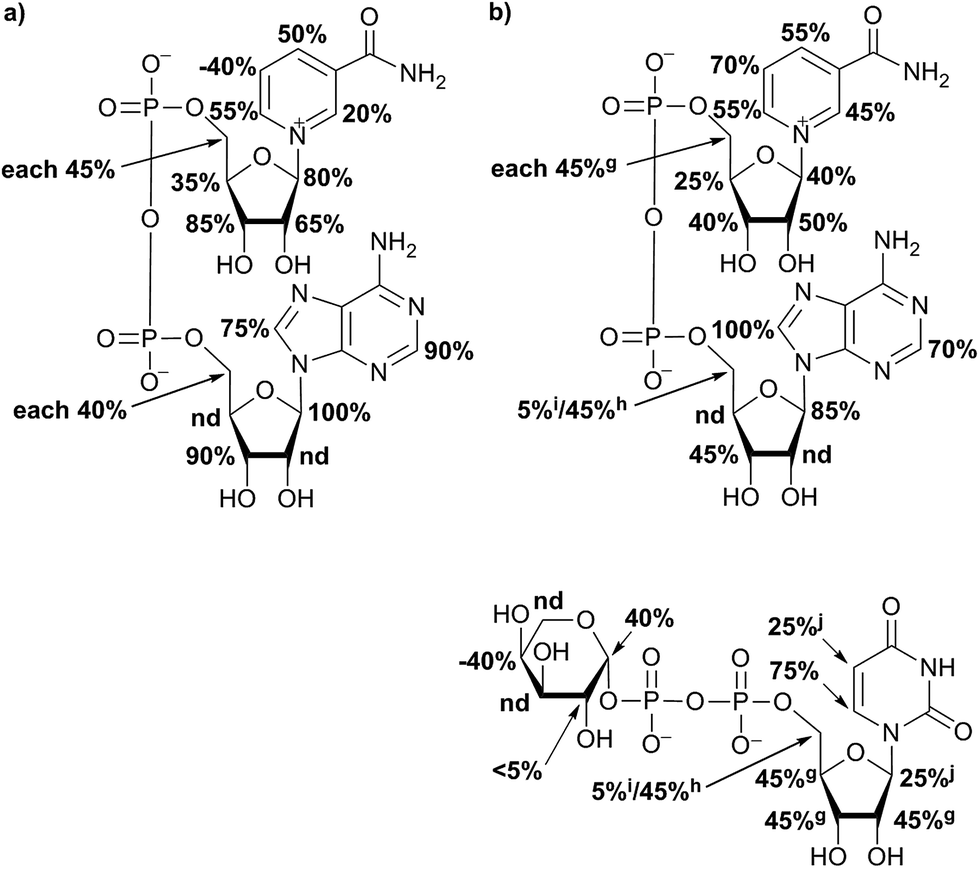 | ||
| Fig. 4 Relative STD effects of co-factor NAD+ without substrate and product showed a strong interaction of aglycone moiety with the protein. Proton at position N5 had a negative STD effect (a). Epitope mapping of NAD+ in presence of UDP-Xyl possessed changes in interaction strength of NAD+ (b). In case of overlapping 1H NMR signals, STD effects are given as equal portion of combined interaction strength and indicated with symbols. | ||
Further, aglycone moieties of NAD+ had moderate to strong STD signal intensities. Only proton at N5 of nicotinamide residue possessed a negative STD effect when no UDP-glycoside was present. This circumstance is very likely caused by water molecule coordination to Thr118, which is located close to the ligand proton.10 However, this water molecule interferes with the ligand during saturation and spin lock event and hence led to a negative STD effect.18
In general, investigation of NAD+ binding to hUXS1A in presence of UDP-Xyl led to higher saturation transfer, suggesting increased contact with groups on the protein. Furthermore, magnetization of co-factor generated some significant changes in STD effects, which are shown in Fig. 4. The strongest change in binding pattern occurred at position N5, probably caused by displacement of D2O for the benefit of an UDP-Xyl moiety.10 These variations in STD effects indicated a complementary interaction between co-factor and UDP-glycosides during binding to hUXS1A. In addition, clearly increased STD response of both aglycone parts in presence of UDP-Xyl, let suggest an approximation of these moieties to the reaction center of hUSX1A (Fig. 5). However, binding pattern of both NAD+ riboses in presence of UDP-Xyl remained similar compared to those of sole NAD+.
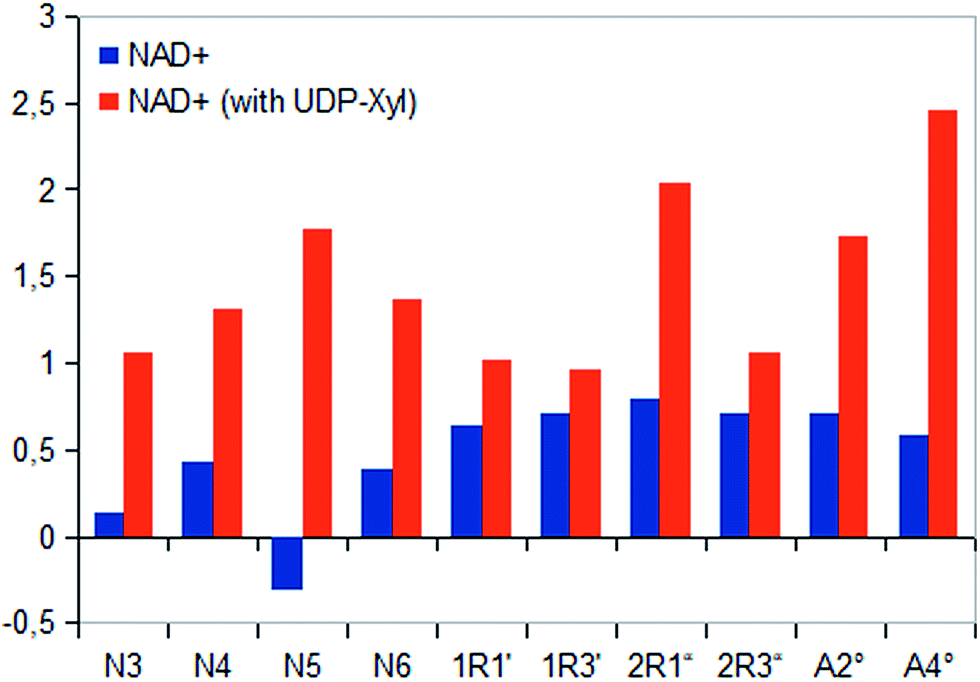 | ||
| Fig. 5 Saturation transfer of sole NAD+ and in presence of UDP-Xyl. Due to varying co-factor concentration absolute STD effects are presented as amplification factors for given saturation time. All protons of co-factor showed increased STD response in existence of enzymatic product. | ||
During catalytic procedure co-factor NAD+ is reduced to NADH and re-oxidized to NAD+.10,13,14 We hence checked if temporary present NADH was released and rebound during the catalytic process. In situ NMR only showed weak increasing signal intensity of one NADH proton close to signal to noise level. However, further proton identification of reduced co-factor was somewhat challenging due to signal overlapping of NAD+ and UDP-GlcUA, respectively UDP-Xyl. Nevertheless, closer consideration of recorded in situ STD spectra gave no evidence for NADH signals (Fig. 6), which would indicate release of NADH in larger amounts. These findings support previous published results10 and demonstrate that NADH is not regularly released from the complex during catalytic event before re-oxidizing to NAD+. Small amounts of potential free NADH rather demonstrated a rare release of reduced co-factor, which is not rebound again and accumulated in solution.
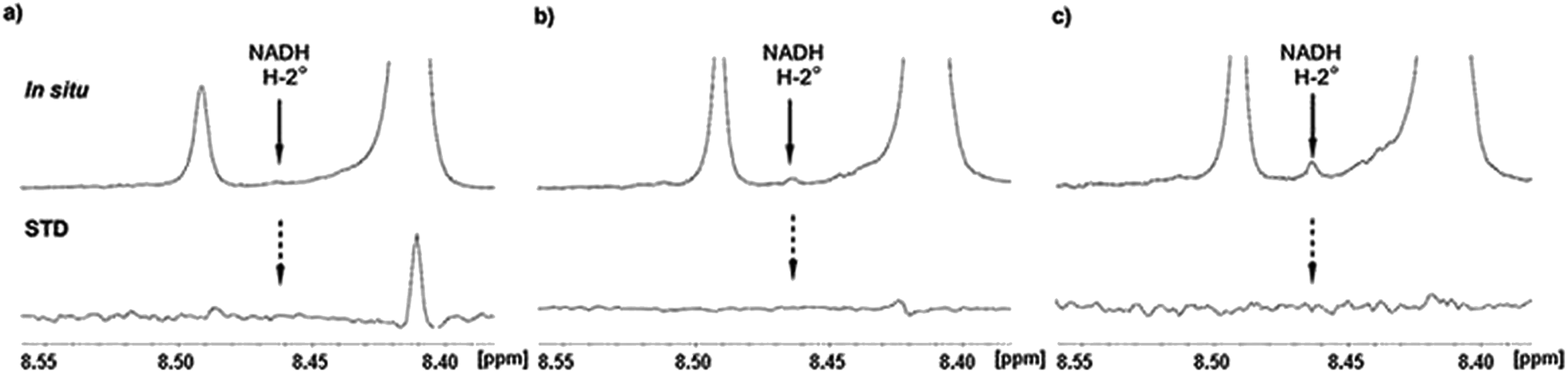 | ||
| Fig. 6 Details from in situ NMR and STD NMR spectra of hUXS1A catalyzed UDP-GlcUA conversion to UDP-Xyl. (a) NMR spectrum ap. 10 min after addition of hUXS1A. No NADH signal of A-2° can be detected neighbored to H-2° of NAD+ (8.41 ppm) and impurity peak (8.49 ppm). Also no STD effect of NADH proton was visible, while A-2° of NAD+ showed an intense STD effect. (b) Reaction progress after 12 h, when 40% of UDP-GlcUA had been transformed. Probably, small amounts of NADH had been accumulated. Also still no STD effect of NADH can be detected. In addition, STD effect on A-2° of NAD+ is decreased, presumably caused by slow denaturation of protein. (c) Potential NADH signal in the in situ1H NMR spectrum after 20 h indicated an accumulation of small amounts during reaction. The released NADH is likely discharged from denatured enzyme. | ||
2.3. Binding pattern of substrate and product during transformation
In situ STD NMR investigations of hUXS1A catalyzed UDP-GlcUA transformation also showed some differences in binding pattern compared to those of UDP-GlcUA measured in absence of external NAD+ (Fig. 7 and 8). In particular, all protons of UDP-GlcUA achieved higher magnetization during catalytic event. Further, comparison of STD response of sole UDP-GlcUA and during transformation, demonstrated clearly increased contact of pyranoside moiety with active site of hUXS1A. These results let suggest a tighter anchorage and better fitting of this area during enzymatic conversion. Simultaneous, decreasing saturation transfer of uracil residue showed reduced interaction with the binding pocket.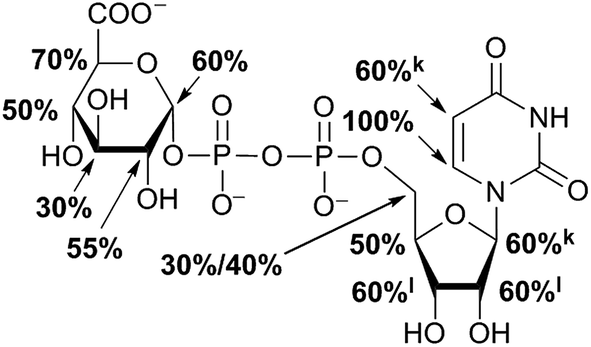 | ||
| Fig. 7 Epitope mapping of substrate during catalytic event. In case of STD signal overlapping, relative STD effects are given as equal portion of combined interaction strength and indicated with superscript symbols. | ||
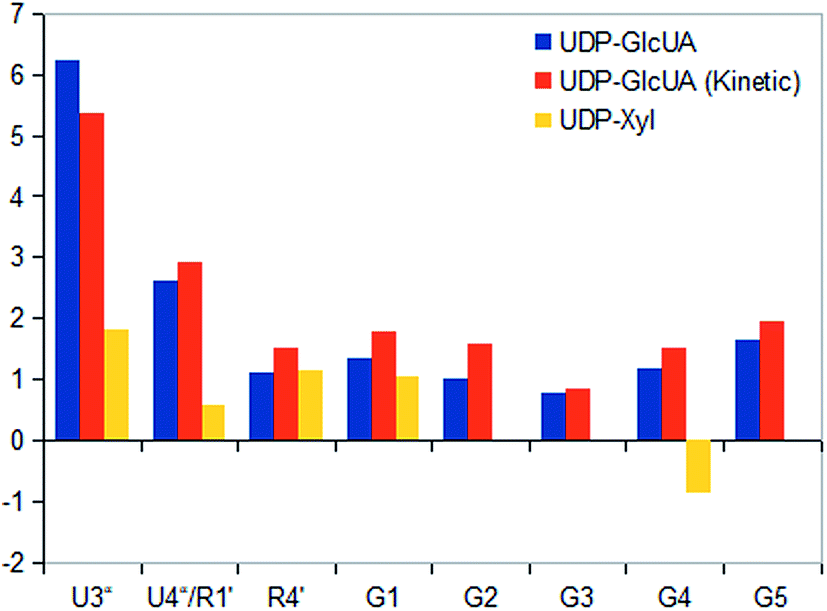 | ||
| Fig. 8 STD responses of sole UDP-GlcUA respectively during enzymatic conversion and of product UDP-Xyl. Absolute saturation transfer is displayed as amplification factor for selected protons due to different substrate concentration. | ||
Enzymatic conversion of UDP-GlcUA to UDP-Xyl is carried out via intermediate UDP-Xyl-4O. We hence checked if small amounts of this intermediate were present in reaction solution during in situ NMR measurements. For this propose, the signal of anomeric proton in xylopyranoside residue of UDP-Xyl-4O displayed the most promising signal based on its isolated and not overlapping position with other present saccharides protons. Nevertheless, precise interpretation was somewhat difficult, as the signal is split into a doublet of doublets and thus challenging to separate from background signal. However, comparison with previous recorded 1H NMR spectra of UDP-Xyl-4O10 and interpretation of in situ NMR and in situ STD NMR measurements showed no observable peaks of this intermediate.4,13 This behavior indicated that UDP-Xyl-4O is not released from hUXS1A during reaction cascade, comparable to NADH.
STD NMR analysis of UDP-Xyl indicated a distinct varying STD response compared to natural substrate UDP-GlcUA. In general, enzymatic product received clearly less magnetization transfer (Fig. 8), suggesting lower affinity to the enzyme. Furthermore, epitope mapping of generated UDP-Xyl possessed an entire different binding pattern compared to UDP-GlcUA, which is shown in Fig. 4. Only proton at G1 in xylopyranoside moiety had a reasonable positive STD effect, while proton G4 showed a large negative STD signal, likely caused by a D2O molecule in a trapped position, leading to interferences with the ligand during saturation and spin lock event.18 This different binding behavior indicated UDP-Xyl to be easily discharged from the enzyme after formation. It also cannot efficiently compete with UDP-GlcUA for binding in the active site and is hence not an effective inhibitor. Such differences between binding of substrate and product enhances the selectivity in forming productive complexes and might lead to higher selectivity and productivity of this enzyme.
2.4. Binding behavior substrate analogues
STD NMR measurements of six different α-glycosides were accomplished. α-methyl-D-glucoside (α-Glc-1-Me), α-isopropyl-D-glucoside (α-Glc-1-iPr), α-methyl-D-xyloside (α-Xyl-1-Me), α-isopropyl-D-xyloside (α-Xyl-1-iPr) as well as α-methyl-D-galactoside (α-Gal-1-Me) and β-methyl-L-arabinoside (β-Ara-1-Me) have a small aglycone part and same glycoside stereochemistry as investigated UDP-sugars. In general, binding patterns were different from those of UDP-sugars and indicated a different mode of action, not guided by anchor groups (Fig. 9). Aglycone moieties of monosaccharides often demonstrated strong STD responses, which might be explained through conformational flexibility of this area, thus receiving more magnetization in contrast to pyranoside protons (Fig. 10). Furthermore, size of aglycone part led to strong variation in saturation transfer and binding pattern. Monosaccharides, having a methyl group as aglycone residue, received more saturation in comparison to corresponding isopropyl-glycosides. These results let assume reduced fitting of synthesized saccharides carrying more space-filling aglycone parts. Moreover, binding patterns of methyl-glycosides differs from corresponding isopropyl-saccharides, suggesting diverse binding modes. Nevertheless, comparison of binding strength and affinity of different carbohydrates to hUXS1A was not enable due to lacking KD values.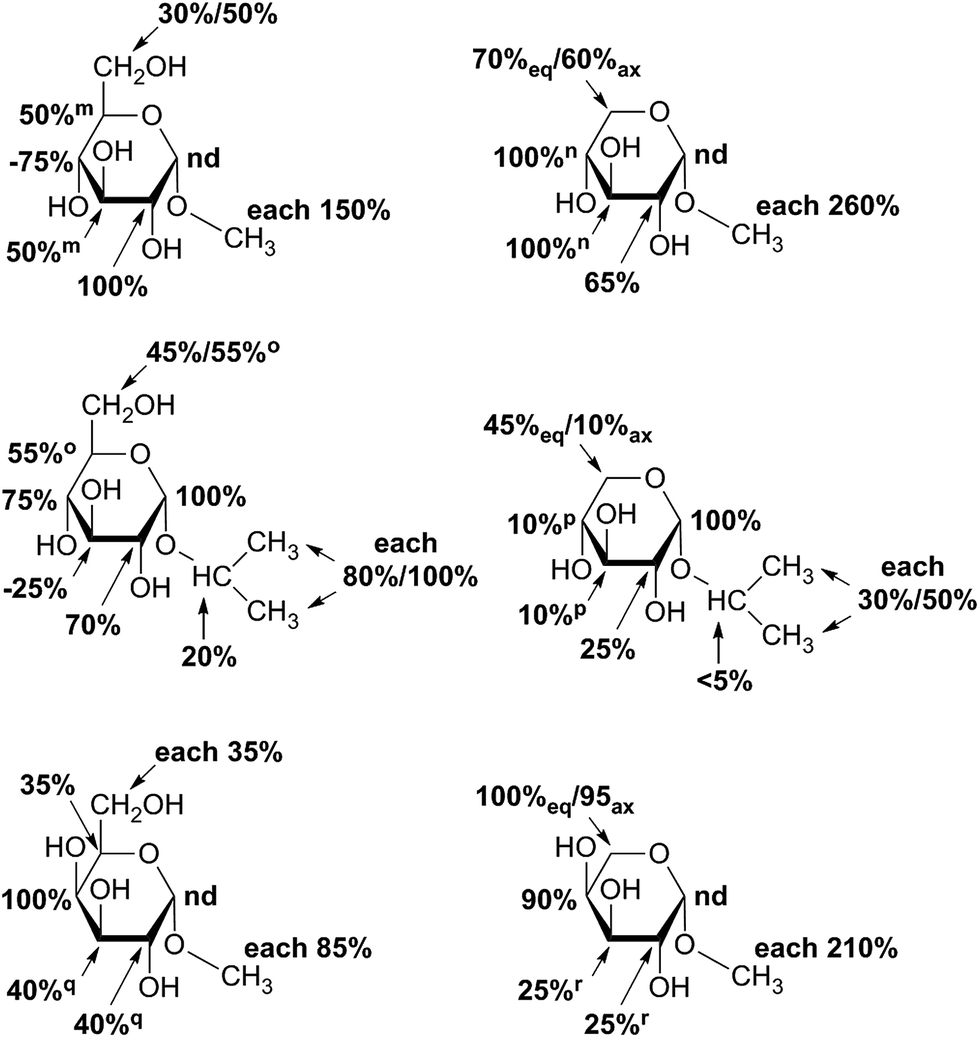 | ||
| Fig. 9 Epitope mapping of investigated monosaccharides bound to hUXS1A in absence of NAD+. In case of signal overlapping in resulting STD spectra, relative effects are given as equal portion of combined interaction strength and indicated with symbols. Proton signals close to the water signal were not determined and denoted as “nd”. Further, axial methylene group proton at G5 is additionally marked with “ax”, respectively equatorial proton is denoted with “eq”. | ||
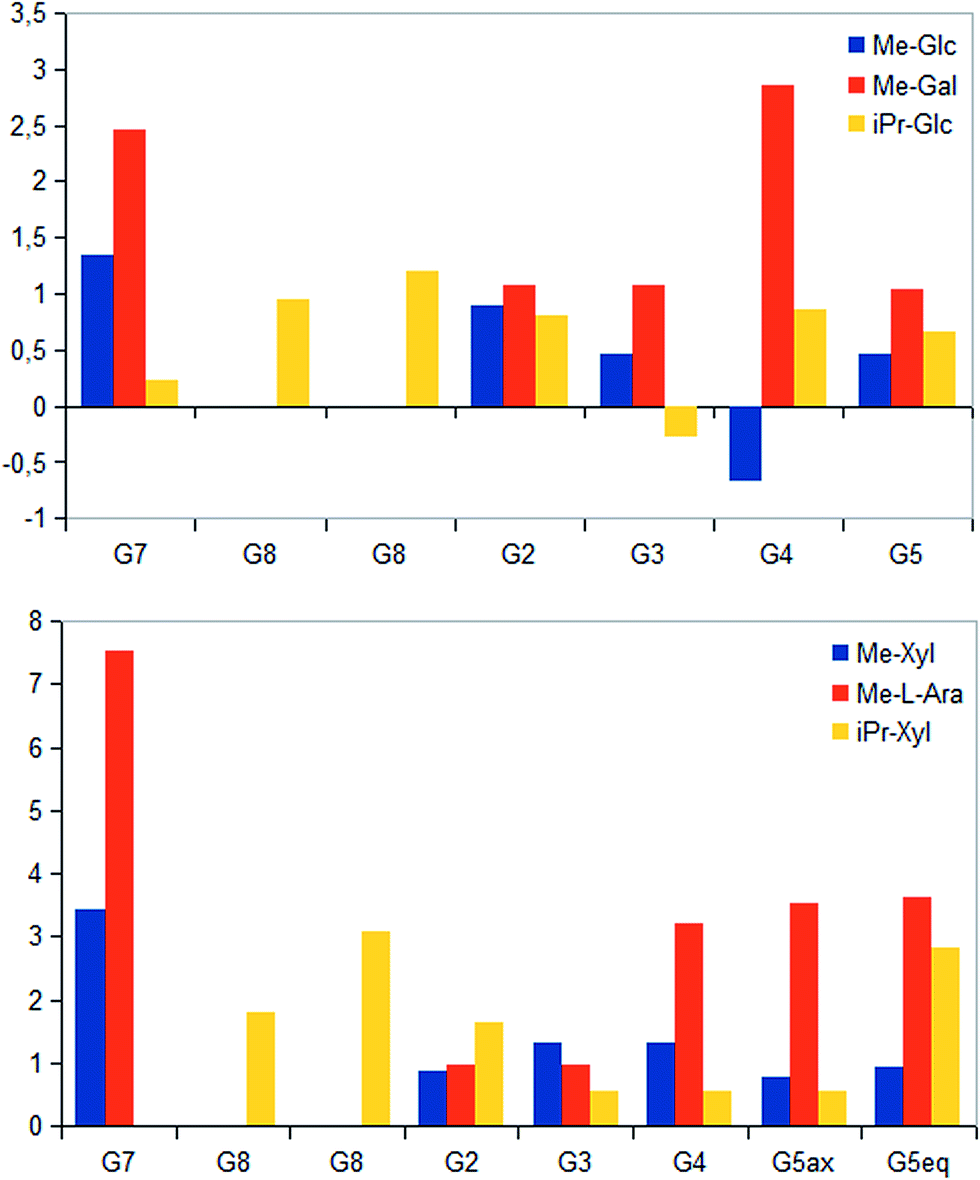 | ||
| Fig. 10 Absolute magnetization transfer for glycoside protons are displayed as amplification factors due to varying saccharide concentration. Absolute STD values were determined as ratios between peak integrals of the STD spectra and peak integrals of the off-resonance spectra. Further, axial methylene group proton at G5 is additionally marked with “ax”, respectively equatorial proton is denoted with “eq”. | ||
Further, both C4 epimers of D-glucose respectively D-xylose possessed some similarities in binding pattern. In particular, proton at G4 in α-Gal-1-Me and β-Ara-1-Me showed a strong STD response in obtained STD NMR spectra. However, due to a clearly different aglycone residue in contrast to UDP-glycosides, no statement can be made concerning potential interaction of proton G4 with Tyr147.
Last, equatorial proton of methylene group at G5 in α-Xyl-1-Me, α-Xyl-1-iPr and β-Ara-1-Me possessed a slightly higher STD response than corresponding axial proton, displaying more intimate contact with groups on the enzyme.
3. Experimental
3.1. Materials
All reagents and solvents were purchased from Sigma-Aldrich (Taufkirchen, Germany) and Alfa Aesar (Karlsruhe, Germany). UDP-saccharides were bought from Carbosynth (Compton, United Kingdom) and deuterated solvents were purchased from Eurisotop (Saint-Aubin, France). All materials not commercially available were synthesized as reported earlier.21 Enzyme stock solution of wild type hUXS1A was prepared and purified as described before.103.2. NMR experiments
NMR samples were prepared in a total volume of 0.70 mL deuterated (99.96%) phosphate buffer (50 mM, pH 7.1).22 They contained a saccharide concentration between 3.3 mM and 13 mM, NAD+ (3.3 mM) and a protein concentration of 6.6 μM, causing a 500–2000 fold excess of investigated ligands. All spectra were obtained at 298.15 K with a Bruker AV III 600 AVANCE spectrometer (Bruker, Rheinstetten, Germany) at 600.13 MHz (1H) using the Bruker Topspin 3.0 software. All experiments were performed in 5 mm high precision NMR sample tubes (Promochem, Wesel, Germany) with a 5 mm PABBO BB probe head. Spectra were referenced to external acetone at 2.225 ppm.STD NMR – for STD NMR measurements pulse program stddiffgp19.3 and the standard program of Topspin 3.0 were applied. All spectra were recorded with a spectral width of 11 ppm and 39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 612 data points. For on-resonance conditions samples were irradiated at −1 ppm. Off-resonance (reference spectrum) irradiation was performed at 30 ppm. Selective saturation of enzyme was achieved by series of Gaussian shaped pulses of 50 ms length with 1 ms delay, giving a total saturation time of 2.0 s. A total number of 256 to 768 scans were recorded, reaching a measurement time of 75 min to 236 min. Corresponding 1H NMR spectra were measured.17 In all spectra WATERGATE was used to suppress the overwhelming HDO signal.23
612 data points. For on-resonance conditions samples were irradiated at −1 ppm. Off-resonance (reference spectrum) irradiation was performed at 30 ppm. Selective saturation of enzyme was achieved by series of Gaussian shaped pulses of 50 ms length with 1 ms delay, giving a total saturation time of 2.0 s. A total number of 256 to 768 scans were recorded, reaching a measurement time of 75 min to 236 min. Corresponding 1H NMR spectra were measured.17 In all spectra WATERGATE was used to suppress the overwhelming HDO signal.23
STD spectra were obtained by subtracting the on-resonance from the corresponding off-resonance spectrum. STD effects were calculated using (I0 − ISTD)/I0, in which the term (I0 − ISTD) defines the peak intensity in the STD spectrum and I0 the peak intensity in the off resonance spectrum. The resulting most intensive STD effect in each spectrum was allocated to 100%. Remaining STD signals were referenced to this most intensive signal.17,18 STD responses of proton signals close to the water signal (+/− 60 Hz) were not taken into account, because of changes in signal intensity caused by water suppression.
Quantification of STD effects were determined using the STD amplification factor (ASTD) for a given saturation time (2.0 s) due to different substrate concentration. ASTD is defined as:16 (I0 − ISTD)/I0 × ligand excess. However, calculation of corrected absolute STD effects via CORCEMA analysis24 was not applicable as not all parameters like kon/koff rates of all compounds influencing the intermolecular saturation transfer were assignable. Hence, detailed comparison of binding strength of different carbohydrates was not enable.
In situ NMR – NMR monitoring of enzymatic reaction was directly accomplished in a NMR tube at 25 °C, initially containing 3.3 mM UDP-GlcUA, 3.3 mM of co-factor NAD+ and 20.8 μM hUXS1A. A series of 24 1H spectra with each 256 scans using WATERGATE suppression were recorded, giving a total observation time of 12 h.
In situ STD NMR – enzymatic transformation was also performed and directly monitored in a NMR tube. STD NMR and 1H NMR were recorded in alternating order during a period of 12 h using same measurement conditions described above. Investigated sample initially comprised 5 mM UDP-GlcUA, 0.5 mM NAD+ and 6.6 μM of hUXS1A.
4. Conclusion and outlook
Obtained STD spectra give significant insights into binding areas of investigated carbohydrates. Location of UDP-saccharides at the active site is guided by the UDP agylcone moiety, showing a quite tight contact to the enzyme. Resulting binding patterns, which are supported by conducted MD simulations of UDP-GlcUA,10 demonstrate a rather flexible and weakly bound pyranoside residue. However, this circumstance enables required pyranoside ring distortion during catalysis. Further, increasing saturation transfer of UDP-GlcUA during catalytic event let suggest a tighter anchorage and better fitting of this area during conversion. Epitope mapping of UDP-Gal demonstrate reduced enzymatic recognition of proton G5 caused by change in stereochemistry at G4. Furthermore, varying binding pattern of product indicate UDP-Xyl to be easily discharged from the enzyme after formation. Likewise, binding studies of substrate analogues clearly differ from those of UDP-sugars and show a different mode of action, not guided by anchor groups. STD and STD in situ NMR derived binding studies of NAD+ indicate tight interaction between co-factor and hUXS1A. It is regularly not released from enzyme while reduced to NADH. Further, higher STD response of NAD+ in presence of UDP-Xyl let suggest increased contact with groups on the protein.Taken together, the analytical methods used can provide data about these binding patterns, which are a valuable basis for directed protein design with the target to generate enzymes accepting UDP-hexuronic acids other than UDP-GlcUA.
Acknowledgements
We kindly thank Ing. Susanne Felsinger (University of Vienna) for helping in NMR measurements. T. E. and B. N. acknowledge financial support from the Austrian Science Fund FWF (project DK Molecular Enzymology; W901-B05).References
- C. E. Clarkin, S. Allen, N. J. Kuiper, B. T. Wheeler, C. P. Wheeler-Jones and A. A. Pitsillides, J. Cell. Physiol., 2011, 226, 749 CrossRef PubMed.
- S. Egger, A. Chaikuad, K. L. Kavanagh, U. Oppermann and B. Nidetzky, Biochem. Soc. Trans., 2010, 38, 1378 CrossRef CAS PubMed.
- J. Y. Lee and A. P. Spicer, Curr. Opin. Cell Biol., 2000, 12, 581 CrossRef CAS PubMed.
- M. Bar-Peled, C. L. Griffith and T. L. Doering, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12003 CrossRef CAS PubMed.
- S. D. Breazeale, A. A. Ribeiro and C. R. H. Raetz, J. Biol. Chem., 2002, 277, 2886 CrossRef CAS PubMed.
- Q. Du, P. Wei, J. Tian, B. Li and D. Zhang, PLoS One, 2013, 8, e60880 CAS.
- M. Mølhøj, R. Verma and W. D. Reiter, Plant J., 2003, 35, 693 CrossRef.
- X. Gu, J. Glushka, Y. Yin, Y. Xu, T. Denny, J. Smith, Y. Jiang and M. Bar-Peled, J. Biol. Chem., 2010, 285, 9030 CrossRef CAS PubMed.
- M. J. Coyne, M. C. Fletcher, B. Reinap and L. E. Comstock, J. Bacteriol., 2011, 193, 5252 CrossRef CAS PubMed.
- T. Eixelsberger, S. Sykora, S. Egger, M. Brunsteiner, K. L. Kavanagh, U. Oppermann, L. Brecker and B. Nidetzky, J. Biol. Chem., 2012, 287, 31349 CrossRef CAS PubMed.
- H. Y. Hwang and H. R. Horvitz, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14218 CrossRef CAS PubMed.
- K. L. Kavanagh, H. Jörnvall, B. Persson and U. Oppermann, Cell. Mol. Life Sci., 2008, 65, 3895 CrossRef CAS PubMed.
- J. S. Schutzbach and D. S. Feingold, J. Biol. Chem., 1970, 245, 2476 CAS.
- H. Ankel and D. S. Feingold, Biochemistry, 1965, 4, 2468 CrossRef CAS.
- S. J. Polizzi, R. M. Walsh, W. B. Peeples, J.-M. Lim, L. Wells and Z. A. Wood, J. Biochem., 2012, 51, 8844 CrossRef CAS PubMed.
- B. Meyer and T. Peters, Angew. Chem., Int. Ed., 2003, 42, 864 CrossRef CAS PubMed.
- B. Meyer and M. Mayer, Angew. Chem., Int. Ed., 1999, 38, 1784 CrossRef.
- L. Brecker, G. D. Straganz, C. E. Tyl, W. Steiner and B. Nidetzky, J. Mol. Catal. B: Enzym., 2006, 42, 85 CrossRef CAS.
- L. Brecker, A. Schwarz, C. Gödl, R. Kratzer, C. E. Tyl and B. Nidetzky, Carbohydr. Res., 2008, 343, 2153 CrossRef PubMed.
- L. Brecker and D. W. Ribbons, Trends Biotechnol., 2000, 18, 197 CrossRef CAS PubMed.
- A. Medgyes, E. Farkas, A. Liptak and V. Pozsgay, Tetrahedron, 1997, 53, 4159 CrossRef CAS.
- pH values were calculated according to: pD = pH + 0.4. P. R. Mussini, T. Mussini and S. Rondinini, Pure Appl. Chem., 1997, 69, 1007 CrossRef CAS.
- M. Piotto, V. Saudek and V. Sklenar, J. Biomol. NMR, 1992, 2, 661 CrossRef CAS PubMed.
- V. Jayalakshmi and N. R. Krishna, J. Am. Chem. Soc., 2005, 127, 14080 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |