DOI:
10.1039/C5RA18257C
(Paper)
RSC Adv., 2015,
5, 93274-93282
Enhancing thermo-stability to ethylene polymerization: synthesis, characterization and the catalytic behavior of 1-(2,4-dibenzhydryl-6-chlorophenylimino)-2-aryliminoacenaphthylnickel halides†
Received
7th September 2015
, Accepted 25th October 2015
First published on 26th October 2015
Abstract
A series of 1-(2,4-dibenzhydryl-6-chlorophenylimino)-2-aryliminoacenaphthylene derivatives (L1–L5) was synthesized and reacted with nickel halides to form the corresponding nickel complexes LNiX2 (X = Br, Ni1–Ni5; X = Cl, Ni6–Ni10). The molecular structures of representative complexes Ni2 and Ni5 were determined by single crystal X-ray diffraction, indicating the distorted square planar geometry around the nickel atom of complex Ni2 and the distorted tetrahedral geometry around the nickel atom of complex Ni5, respectively. Upon activation with low amounts of ethylaluminium sesquichloride (Et3Al2Cl3, EASC), all nickel complexes exhibited high activities up to 1.09 × 107 g of PE per mol of Ni per h toward ethylene polymerization, producing branched polyethylenes. Most importantly, these systems showed good thermo-stability, even at 80 °C maintaining the activity with 3.76 × 106 g of PE per mol of Ni per h.
Introduction
Two decades have passed since the discovery of α-diiminonickel pre-catalysts in ethylene polymerization,1 and the rapid progress of the nickel complex pre-catalysts has been considered timely.2 Regarding the foremost model of α-diiminonickel pre-catalysts (A, Scheme 1),1,3 the catalytic activities of their nickel complexes were enhanced by using the bulky 2,3-bis(2,6-diarylphenylimino)butanes4 as well as dibenzhydryl-substituted 2,3-diiminobutanes,5 in addition, the good thermo-stable precatalysts were achieved.5c In parallel, the dibenzhydryl-substituted-1,2-bis(arylimino)acenaphthylene derivatives have also been developed and used to improve the catalytic activities of their nickel complexes (B, Scheme 1);6–8 besides higher activities achieved, the properties of resultant polyethylenes were tailored through changing substituents from methyl,6 chloro-7 and fluoro-group8 with producing narrow polydispersion polyethylenes from low to high molecular weights. It is still far to meet the industrial demands regarding the thermo-stable precatalysts, fortunately the 1-(2,4-dibenzhydryl-6-methylphenylimino)-2-aryliminoacenaphthyl nickel complexes performed high ethylene polymerization at high temperature (C, Scheme 1)9a along with extensive analogous complexes.9b,9c The computational study clearly indicated the positive influence of the electron-withdrawing substituents to the late-transition metal precatalysts,10 which were consistent to experimental observations.5a,7,8 Instead of 2,4-dibenzhydryl-6-methylphenylamine9a by 2,4-dibenzhydryl-6-chlorophenylamine, subsequently the 1-(2,4-dibenzhydryl-6-chlorophenylimino)-2-aryliminoacenaphthylene derivatives were prepared and reacted with nickel halides to form the title nickel complexes (D, Scheme 1). These new nickel complexes exhibited high activities up to 1.09 × 107 g of PE per mol of Ni per h for ethylene polymerization at 50 °C in comparison to its analogs at 20 °C,9a more importantly, the system maintained good thermal stability and high activity with 3.76 × 106 g of PE per mol of Ni per h up to 80 °C. Therefore the title complex precatalysts would be potentially useful in industrial process, and further investigations remain to be interesting. The resulting polyethylenes possessed high molecular weights, but narrow polydispersity. The syntheses and characterization of the nickel complexes are reported in detail as well as their catalytic behavior toward ethylene polymerization, and the properties of the resultant polyethylenes were explored with regard to the molecular weight, polydispersity as well as branching.
 |
| | Scheme 1 α-Diiminonickel complex precatalysts. | |
Results and discussions
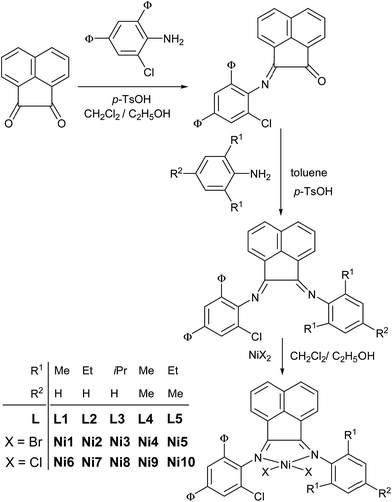
Synthesis of 1-(2,4-dibenzhydryl-6-chlorophenylimino)-2-arylimino acenaphthylenes (L1–L5) and their nickel complexes (Ni1–Ni10)
The stoichiometric condensation of acenaphtylene-1,2-dione with 2,4-dibenzhyldryl-6-chlorophenylamine was conducted to form the 2-(2,4-dibenzhydryl-6-chlorophenylimino)acenaphthylenone, which further reacted with various anilines to afford the 1-(2,4-dibenzhydryl-6-chlorophenylimino)-2-arylimino-acenaphthylene derivatives (L1–L5, Scheme 2). Then the routine reactions of these organic ligands (L1–L5) with either (DME)NiBr2 (DME as dimethoxyethane) or NiCl2·6H2O formed their corresponding nickel complexes as bromides Ni1–Ni5 or chlorides Ni6–Ni10 (Scheme 2) in acceptable yields, respectively. Reflected by the FT-IR spectra, the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N of the nickel halide complexes Ni1–Ni10 showed weaker intensities in the range of 1660–1630 cm−1 in comparing with the range of 1670–1650 cm−1 for the free organic compounds L1–L5, indicating effective coordination between cationic nickel and Nimino atom. To confirm the absolute structure, the single crystals of representative complexes Ni2 and Ni5 were obtained and characterized by the single crystal X-ray diffraction.
N of the nickel halide complexes Ni1–Ni10 showed weaker intensities in the range of 1660–1630 cm−1 in comparing with the range of 1670–1650 cm−1 for the free organic compounds L1–L5, indicating effective coordination between cationic nickel and Nimino atom. To confirm the absolute structure, the single crystals of representative complexes Ni2 and Ni5 were obtained and characterized by the single crystal X-ray diffraction.
 |
| | Scheme 2 Synthesis of ligands L1–L5 and their nickel complexes Ni1–Ni10. | |
X-ray crystallography
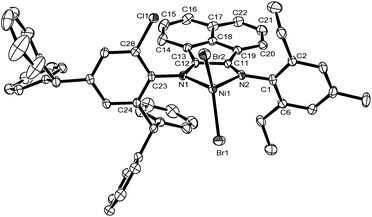
Single crystals of complexes Ni2 and Ni5 were obtained by layering diethyl ether onto their dichloromethane solution, respectively. There are two independent molecules of the complex Ni2 together with free dichloromethane in the crystal cell adapting the distorted square planar geometry around the nickel, shown in Fig. 1. The complex Ni5 described the distorted tetrahedral geometry around the nickel, shown in Fig. 2. The different configurations of complexes Ni2 and Ni5 may be caused by substituents and the solvent molecules in the crystal cells. Their selected bond lengths and angles are tabulated in Table 1.
 |
| | Fig. 1 ORTEP drawing of Ni2 with thermal ellipsoids at a 30% probability level. Hydrogen atoms and molecule of dichloromethane have been omitted for clarity. | |
 |
| | Fig. 2 ORTEP drawing of Ni5 with thermal ellipsoids at a 30% probability level. Hydrogen atoms have been omitted for clarity. | |
Table 1 Selected bond lengths (Å) and angles (°) for complexes Ni2 and Ni5
| Ni2 |
Ni5 |
| Bond lengths (Å) |
| Ni(1)–Br(1) |
2.5077(6) |
Ni(1)–Br(1) |
2.3172(8) |
| Ni(1)–Br(2) |
2.4978(6) |
Ni(1)–Br(2) |
2.3360(8) |
| Ni(1)–N(1) |
2.158(3) |
Ni(1)–N(1) |
2.031(3) |
| Ni(1)–N(2) |
2.147(3) |
Ni(1)–N(2) |
2.010(3) |
| N(1)–C(12) |
1.272(4) |
N(1)–C(12) |
1.277(5) |
| N(1)–C(23) |
1.409(4) |
N(1)–C(23) |
1.435(4) |
| N(2)–C(11) |
1.272(5) |
N(2)–C(11) |
1.282(5) |
| N(2)–C(1) |
1.411(4) |
N(2)–C(1) |
1.440(5) |
| Cl(1)–C(28) |
1.729(4) |
Cl(1)–C(28) |
1.724(4) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Bond angles (°) |
| N(2)–Ni(1)–N(1) |
79.52(11) |
N(2)–Ni(1)–N(1) |
82.70(13) |
| Br(1)–Ni(1)–Br(2) |
102.34(2) |
Br(1)–Ni(1)–Br(2) |
125.62(3) |
| N(2)–Ni(1)–Br(1) |
165.66(8) |
N(2)–Ni(1)–Br(1) |
105.90(10) |
| N(1)–Ni(1)–Br(1) |
89.20(8) |
N(1)–Ni(1)–Br(1) |
114.83(9) |
| N(2)–Ni(1)–Br(2) |
88.94(7) |
N(2)–Ni(1)–Br(2) |
115.66(10) |
| N(1)–Ni(1)–Br(2) |
168.43(8) |
N(1)–Ni(1)–Br(2) |
104.13(9) |
According to Fig. 1, the square plane was formed with the N1, N2, Br2 and Br1 atoms and the nickel atom having a deviation distance of 0.090 Å from the plane. The bond length of Ni1–N1 (2.158 Å) is slightly longer than the value of corresponding Ni1–N2 (2.147 Å), indicating the steric hindrance of ligands and being consistent with their analogues.6–9 The typical CN double-bond character were observed within bonds N1–C12 (1.272 Å) and N2–C11 (1.272 Å), being significantly shorter than the typical C–N single bonds of the N1–C23 (1.409 Å) and N2–C1 (1.411 Å). Moreover, the Ni–Br bond lengths are 2.5077 Å (Ni1–Br1) and 2.4978 Å (Ni1–Br2).
Complex Ni5, with the distorted tetrahedral geometry around the nickel center being consistent to its analogous complexes,6,9a has the basal plane of the N1, N2 and Br1 atoms with the apical atom of the Br2. The distance between nickel atom and the basal plane is 0.939 Å. The dihedral angle between the plane formed by N1, N2 and Ni1, and the basal plane is 38.26°. The Ni–N bond lengths are 2.031 Å (Ni1–N1) and 2.010 Å (Ni1–N2). Similar to the complex Ni2, the bonds N1–C12 (1.277 Å) and N2–C11 (1.282 Å) have the typical CN double-bond character, and are shorter than the single-bond N1–C23 (1.436 Å) and N2–C1 (1.441 Å). The Ni–Br bond lengths are 2.317 Å (Ni1–Br1) and 2.336 Å (Ni1–Br2). The dihedral angles between the chelate plane including N1, N2 and Ni1 and N1-aryl ring or N2-aryl ring are 82.64° and 77.43°, respectively.
Ethylene polymerization
Complex Ni1 was used to optimize the ethylene polymerization parameters to find a suitable co-catalyst. The ethylene polymerization trials were conducted through using various alkylaluminium reagents such as methylaluminoxane (MAO), modified methylaluminoxane (MMAO), dichloride ethylaluminium (AlEtCl2) and ethylaluminium sesquichloride (Et3Al2Cl3, EASC) as activators at 30 °C under 10 atm ethylene; their results are summarized in Table 2. In general, the Ni1/EASC system exhibited the highest activity toward ethylene polymerization (entries 1–4, Table 2). Therefore further extensive investigations were explored with EASC as co-catalyst.
Table 2 Selection of a suitable co-catalyst based on Ni1a
| Entry |
Co-cat. |
Al/Ni |
Act.b |
Tmc/°C |
Mwd |
Mw/Mnd |
| Conditions: 2.0 μmol of Ni1; 10 atm of ethylene; 30 °C; 30 min; 100 mL toluene. 106 g of PE per mol of Ni per h. Determined by DSC. Determined by GPC, 104 g mol−1. |
| 1 |
MAO |
3000 |
2.64 |
120.5 |
33.36 |
2.3 |
| 2 |
MMAO |
3000 |
1.25 |
131.0 |
2.67 |
1.9 |
| 3 |
EtAlCl2 |
500 |
1.97 |
131.4 |
5.85 |
2.5 |
| 4 |
EASC |
500 |
5.68 |
122.2 |
18.51 |
2.6 |
In presence of EASC, the complex Ni1 was explored to optimize the polymerization parameters including the Al/Ni molar ratio, reaction temperature and reaction period (Table 3). At 20 °C, the catalytic activities gradually increased with variations of the Al/Ni molar ratio from 200 to 500 (entries 1–4, Table 3), then slightly decreased with further higher Al/Ni molar ratio as 600 (entry 5, Table 3), indicating the optimum Al/Ni ratio as 500.
Table 3 Ethylene polymerization by Ni1–Ni10/EASCa
| Entry |
Pre-cat. |
Al/Ni |
T/°C |
t/min |
Act.b |
Tmc/°C |
Mwd |
Mw/Mnd |
| Conditions: 2.0 μmol of Ni1; ethylene pressure 10 atm; total volume 100 mL. 10 6 g of PE per mol of Ni per h. Determined by DSC. Determined by GPC, 104 g mol−1. Ethylene pressure 1 atm. Ethylene pressure 5 atm. |
| 1 |
Ni1 |
200 |
20 |
30 |
1.87 |
131.7 |
35.72 |
2.3 |
| 2 |
Ni1 |
300 |
20 |
30 |
4.03 |
124.8 |
24.55 |
3.1 |
| 3 |
Ni1 |
400 |
20 |
30 |
4.19 |
125.0 |
24.47 |
3.6 |
| 4 |
Ni1 |
500 |
20 |
30 |
4.49 |
123.0 |
25.11 |
3.3 |
| 5 |
Ni1 |
600 |
20 |
30 |
3.77 |
124.5 |
26.69 |
3.3 |
| 6 |
Ni1 |
500 |
30 |
30 |
5.68 |
122.2 |
18.51 |
2.6 |
| 7 |
Ni1 |
500 |
40 |
30 |
7.83 |
107.6 |
8.07 |
2.4 |
| 8 |
Ni1 |
500 |
50 |
30 |
10.86 |
101.0 |
5.43 |
2.5 |
| 9 |
Ni1 |
500 |
60 |
30 |
10.30 |
79.2 |
2.90 |
1.8 |
| 10 |
Ni1 |
500 |
70 |
30 |
9.24 |
79.2 |
2.07 |
1.7 |
| 11 |
Ni1 |
500 |
80 |
30 |
3.76 |
72.4 |
0.75 |
2.5 |
| 12 |
Ni1 |
500 |
50 |
15 |
16.48 |
88.9 |
4.74 |
2.1 |
| 13 |
Ni1 |
500 |
50 |
45 |
9.17 |
98.0 |
6.46 |
2.5 |
| 14 |
Ni1 |
500 |
50 |
60 |
8.69 |
111.6 |
7.70 |
2.0 |
| 15 |
Ni2 |
500 |
50 |
30 |
8.07 |
96.7 |
7.97 |
2.4 |
| 16 |
Ni3 |
500 |
50 |
30 |
6.62 |
59.9 |
8.78 |
2.1 |
| 17 |
Ni4 |
500 |
50 |
30 |
8.54 |
97.0 |
4.71 |
2.2 |
| 18 |
Ni5 |
500 |
50 |
30 |
7.88 |
83.1 |
6.91 |
2.1 |
| 19 |
Ni6 |
500 |
50 |
30 |
6.47 |
98.7 |
5.55 |
2.2 |
| 20 |
Ni7 |
500 |
50 |
30 |
5.62 |
97.4 |
9.41 |
2.3 |
| 21 |
Ni8 |
500 |
50 |
30 |
3.11 |
62.6 |
9.42 |
2.2 |
| 22 |
Ni9 |
500 |
50 |
30 |
6.05 |
107.2 |
5.97 |
2.4 |
| 23 |
Ni10 |
500 |
50 |
30 |
3.54 |
96.5 |
7.28 |
2.6 |
| 24e |
Ni1 |
500 |
50 |
30 |
0.69 |
52.9 |
17.63 |
2.6 |
| 25f |
Ni1 |
500 |
50 |
30 |
4.72 |
95.7 |
8.75 |
2.0 |
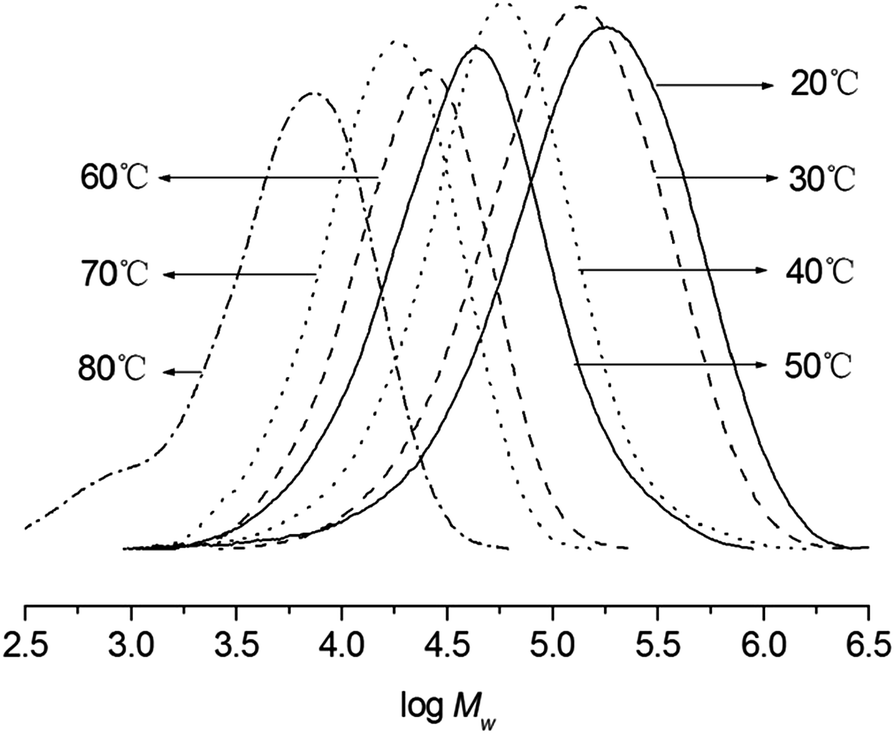
Fixing the Al/Ni ratio at 500, the reaction temperatures were increased from 20 to 80 °C (entries 4, 6–11, Table 3); the highest activity of 1.09 × 107 g of PE per mol of Ni per h was observed at 50 °C (entry 8, Table 3). It is worthily mentioned that the polymerization at 80 °C (entry 11, Table 3) maintained a high activity with 3.76 × 106 g of PE per mol of Ni per h, meaning that the current system possessed better thermal-stability than their analogous pre-catalysts.5a,6,7,9a Regarding the GPC curves of obtained polyethylenes (Fig. 3), the higher molecular weights of polyethylenes were obtained at the lower reaction temperatures (entries 4, 6–11, Table 3), the chain transfer and termination would easily take place at the elevated temperature.
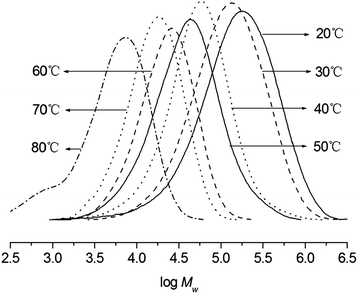 |
| | Fig. 3 GPC curves of polyethylenes by the Ni1/EASC system at different temperature (entries 4, 6–11 in Table 3). | |
Regarding the lifetime of the active species, the ethylene polymerization was conducted over different periods such as 15, 30, 45 and 60 min (entries 8 and 12–14, Table 3). The highest activity was observed within 15 min (entry 12, Table 3), probably reflecting that the active species were quickly formed upon the addition of EASC. The catalytic activities gradually decreased with prolonging the reaction time, meanwhile the resultant polyethylene possessed the higher molecular weight along with a longer reaction time (Fig. 4), indicating some catalytic species remained active11 and consistent with the literature observations.6,7,9a,12
 |
| | Fig. 4 GPC curves of polyethylenes by the Ni1/EASC system at different times (entries 8 and 12–14 in Table 3). | |
Regarding the ethylene pressure, the polymerization experiments were conducted under different pressures such as the 1 atm, 5 atm and 10 atm (entries 8 and 24–25 in Table 3). Higher molecular weights of the resulting polyethylenes were achieved under lower ethylene pressures (Fig. 5), which is quite unusual and differing from the common conclusion that the polyethylenes obtained at higher ethylene pressures showed higher molecular weights. Two competitive reactions as chain propagation and migration parallel exist within polymerization of ethylene. The current system results the faster chain migration than propagation at higher pressures, therefore, polyethylenes with lower molecular weights were obtained at higher ethylene pressure, meanwhile the termination and reformation of active species happened. Such phenomena were previously observed also with precatalysts of palladium13a,b and cobalt complexes.13c
 |
| | Fig. 5 GPC curves of polyethylenes by the Ni1/EASC system at different pressures (entries 8 and 24–25 in Table 3). | |
Under the optimum conditions with an Al/Ni ratio of 500 at 50 °C, other bromide complexes were also investigated (entries 15–18 in Table 3) and showed high activities toward ethylene polymerization. The activities decrease in the order Ni1 [2,6-di(Me)] > Ni4 [2,4,6-tri(Me)] > Ni2 [2,6-di(Et)] > Ni5 [2,6-di(Et)-4-Me] > Ni3 [2,6-di(i-Pr)]. Less bulky ortho-substituents (R1 group; Scheme 2) in the complexes, the higher catalytic activities; it was considered of the bulky 2,4-dibenzhydryl-6-chlorophenyl group already occupying some space around nickel site.7,8,9c
Besides the molecular weights and polydispersity measured by the GPC measurement, the DSC data were measured and showed most of the Tm values higher than 100 °C, being generally higher than these of the analogous nickel precatalysts.7,9 The Tm values are usually relative to the branches of polyethylenes: higher Tm value corresponds to lower branched of polyethylene. The Tm values of polyethylenes obtained at 50 and 60 °C (entries 8 and 9, Table 3) were observed as 101.0 and 79.2 °C, and two samples in deuterated 1,2-dichlorobenzene were measured by the 13C NMR measurement. Interpreted according to the literature,14 the polyethylene produced at 50 °C (Fig. 6) possessed 46 branches per 1000 carbons including methyl (62.4%), ethyl (16.8%) and longer chains (20.8%), whilst the polyethylene obtained at 60 °C (Fig. 7) had 59 branches per 1000 carbons, containing methyl (47.1%), ethyl (8.0%) and longer chains (44.9%). At higher polymerization temperature, it is higher potential to have chain termination and migration instead of chain propagation; and the higher possibility of longer branches would be observed due to more chain migration.15 That was consistent to the palladium precatalysts forming polyethylenes with high branching at the higher temeprature.13b Generally the polyethylenes obtained in the current system possessed lower branches than these produced by the nickel analogous precatalysts.7,9
 |
| | Fig. 6 13C NMR spectrum of the polyethylenes obtained with Ni1/EASC at 50 °C (entry 8, Table 3). | |
 |
| | Fig. 7 13C NMR spectrum of the polyethylenes obtained with Ni1/EASC at 60 °C (entry 9, Table 3). | |
Extensively, the nickel chlorides (Ni6–Ni10) were investigated and showed high activities towards ethylene polymerization (entries 19–23, Table 3). The tendency of their catalytic performance was similar to that showed above by their bromide analogs. Comparing two sets of catalytic data by the bromide and chloride complexes, the bromide precatalysts generally performed higher activities than chloride precatalysts did, but chloride precatalysts produced polyethylenes with slightly higher molecular weights; these trends were in good agreement with observations by its analogs.7,8
Conclusion
A series of 1-(2,4-dibenzhydryl-6-chlorophenylimino)-2-aryliminoacenaphthyl nickel halides was prepared and characterized as well as the single crystal X-ray diffraction of representative complexes. Upon activation with EASC at very low Al/Ni ratio (500), all nickel complex pre-catalysts exhibited high activities up to 1.09 × 107 g of PE per mol of Ni per h toward ethylene polymerization; importantly the optimum polymerization temperature was operated at 50 °C along with high activity maintained at 80 °C in the trial of Ni1, indicating the better thermal stability than their analogs. The bromide precatalysts showed higher activities than their chloride analogs did, but the chloride precatalysts produced polyethylenes with higher molecular weights. The branching degrees of polyethylenes were observed as several tens by 13C NMR measurement, which were slightly lower than these obtained by their analogs.7–9 Therefore the polyethylenes could be well tailored in controlling molecular weights and branching through finely tuning the substituents of ligands within 1,2-bis(arylimino)acenaphthylnickel complexes, which are potentially modified to be a practicable precatalysts for a commercial process in near future.
Experimental section
General procedure
All manipulations of air and/or moisture sensitive compounds were carried out under a nitrogen atmosphere using standard Schlenk techniques. All solvents were refluxed and purified under a nitrogen atmosphere before to use. Methylaluminoxane (MAO) (1.46 mol L−1 in toluene) and methylaluminoxane (MMAO) (1.93 mol L−1 in heptane, 3A) were purchased from Akzo Nobel Corp. Dichloride ethylaluminium (AlEtCl2) (1.44 mol L−1 in toluene) and ethylaluminium sesquichloride (Et3Al2Cl3, EASC, 0.87 mol L−1 in toluene) were purchased from Acros Chemical. High-purity ethylene was purchased from Beijing Yanshan Petrochemical Co. and used as received. Other reagents were purchased from Aldrich, Acros, or local suppliers. 1H and 13C NMR spectra were recorded on a Bruker DMX 400 MHz instrument at ambient temperature using TMS as an internal standard; δ values were given in ppm and J values in Hz. FT-IR spectra were recorded on a Perkin-Elmer System 2000 FT-IR spectrometer. Elemental analyses were carried out using a Flash EA 1112 microanalyzer. Molecular weights (Mw) and molecular weight distribution (MWD) of polyethylene were determined by a PL-GPC220 at 150 °C, with 1,2,4-trichlorobenzene as the solvent. The trace and melting points of polyethylene were measured from the second scanning run on Perkin-Elmer TA-Q2000 DSC analyzer under a nitrogen atmosphere. In the procedure, a sample of about 2.0–4.0 mg was heated to 150 °C at a heating rate of 20 °C min−1, and kept for 5 min at 150 °C to remove the thermal history and then cooled at a rate of 20 °C min−1 to −20 °C. 13C NMR spectra of the polyethylene was recorded on a Bruker DMX 300 MHz instrument at 135 °C in deuterated 1,2-dichlorobenzene with TMS as an internal standard.
Syntheses and characterization
The syntheses of 1-(2,4-dibenzhydryl-6-chlorophenylimino)-2-aryliminoacenaphethylene derivatives and their nickel halide complexes were performed according to the literature procedure (Scheme 2). The 2-(2,4-dibenzhydryl-6-chlorophenylimino)acenaphthylenone was firstly prepared by the stoichiometrial condensation of 2,4-dibenylmethyl-6-chloroaniline with acenaphthylene-1,2-dione, and then further reacted with common aniline derivatives to obtain unsymmetrical 1,2-diiminoacenaphthylene derivatives (L1–L5), which individually reacted with either (DME)NiBr2 or NiCl2 to form their bromide (Ni1–Ni5) or chloride complexes (Ni6–Ni10), respectively.
2-(2,4-Dibenzhydryl-6-chlorophenylimino)acenaphthylenone. A mixture of 2,4-diphenylmethyl-6-chloroaniline (18.38 g, 40 mmol), acenaphthylene-1,2-dione (7.28 g, 40 mmol) and a catalytic amount of p-toluenesulfonic acid (1.50 g) were mixed in 500 mL dichloromethane and 50 mL ethanol, and the mixture was stirred at room temperature over night. The solution was concentrated under vacuum, and its residue was further purified by alumina column chromatography (50/1 petroleum ether/ethyl acetate) to obtain the expected compound as the red powder in 9.73 g (39%). Mp: 126–128 °C. 1H NMR (400 MHz, CDCl3, TMS): δ 8.07 (t, J = 7.4 Hz, 2H), 7.88 (d, J = 8.4 Hz, 1H), 7.75 (t, J = 7.6 Hz, 1H), 7.65–7.21 (m, 7H), 7.14–6.98 (m, 7H), 6.96 (d, J = 6.8 Hz, 3H), 6.78 (s, 1H), 6.75 (d, J = 4.0 Hz, 2H), 6.56 (d, J = 7.2 Hz, 1H), 6.47 (t, J = 7.6 Hz, 2H), 6.22 (t, J = 7.4 Hz, 1H), 5.61 (s, 1H), 5.49 (s, 1H). 13C NMR (100 MHz, CDCl3, TMS): δ 144.5, 143.3, 141.0, 135.8, 131.8, 130.3, 129.6, 129.3, 129.2, 128.4, 128.3, 128.1, 127.9, 127.7, 127.6, 127.5, 126.5, 126.3, 125.3, 123.0, 121.8.
1-(2,4-Dibenzhydryl-6-chlorophenylimino)-2-(2,6-dimethyl phenylimino)acenaphthylene (L1). 2.49 g (4.0 mmol) 2-(2,4-dibenzhydryl-6-chlorophenylimino)acenaphethylenone, 0.73 g (6.0 mmol) 2,6-dimethylaniline and a catalytic amount of 0.3 g p-toluenesulfonic acid were mixed and refluxed in 100 mL toluene for 2 h. The solution was concentrated under vacuum, then its residue was further purified by the alumina column chromatography (50/1 petroleum ether/ethyl acetate) to obtain the compound L1 as the yellow powder, 0.46 g (16%). Mp: 138–140 °C. 1H NMR (300 MHz, CDCl3, TMS): δ 7.78 (t, J = 8.4 Hz, 2H), 7.31 (t, J = 7.1 Hz, 5H), 7.23–7.08 (m, 16H), 6.89 (d, J = 7.2 Hz, 2H), 6.76 (s, 1H), 6.59 (d, J = 7.2 Hz, 1H), 6.53–6.46 (m, 3H), 6.27 (t, J = 7.2 Hz, 1H), 5.79 (s, 1H), 5.51 (s, 1H), 2.27 (s, 3H), 2.08 (s, 3H). 13C NMR (100 MHz, CDCl3, TMS): δ 142.6, 141.3, 140.5, 140.4, 136.2, 130.5, 130.0, 129.7, 129.5, 129.4, 129.1, 128.9, 128.8, 128.5, 128.3, 128.2, 128.0, 127.7, 127.5, 126.6, 126.3, 125.5, 125.0, 124.9, 123.9, 123.2, 122.3, 122.2, 56.2, 52.8, 31.7, 22.8, 18.3, 17.9, 14.3. IR (KBr; cm−1): 3025(w), 2957(w), 2920(w), 2361(s), 1738(m), 1669(s), 1643(m), 1595(s), 1552(m), 1494(s), 1444(s), 1234(m), 1206(m), 1078(m), 1036(m), 923(s), 888(m), 698(vs). Anal. calcd for C52H39N2Cl (727.33): C, 85.87; H, 5.40; N, 3.85. Found: C, 85.39; H, 5.80; N, 3.62.
1-(2,4-Dibenzhydryl-6-chlorophenylimino)-2-(2,6-diethyl phenylimino)acenaphethylene (L2). According to the synthetic procedure of L1, the compound L2 was obtained from the reaction of 2-(2,4-dibenzhydryl-6-chlorophenylimino)acenaphthylenone (2.00 g, 3.20 mmol), 2,6-diethylaniline (0.72, 4.80 mmol) and p-toluenesulfonic acid (0.2 g), as the yellow powder in 0.26 g (11%). Mp: 180–182 °C. 1H NMR (400 MHz, CDCl3, TMS): δ 7.77 (t, J = 8.8 Hz, 2H), 7.33–7.27 (m, 5H), 7.24–7.12 (m, 13H), 7.00 (d, J = 6.8 Hz, 3H), 6.89 (d, J = 7.2 Hz, 2H), 6.77 (s, 1H), 6.57 (d, J = 6.8 Hz, 1H), 6.52–6.48 (m, 3H), 6.28 (t, J = 7.2 Hz, 1H), 5.80 (s, 1H), 5.51 (s, 1H), 2.77–2.68 (m, 1H), 2.62–2.49 (m, 2H), 2.41–2.31 (m, 1H), 2.24 (t, J = 7.4 Hz, 3H), 1.06 (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3, TMS): δ 164.1, 163.7, 161.0, 148.4, 143.6, 143.5, 142.6, 141.1, 140.2, 136.1, 130.7, 129.8, 129.6, 129.3, 129.1, 128.8, 128.6, 128.4, 128.0, 127.7, 127.6, 127.4, 126.5, 126.4, 126.2, 125.4, 124.1, 123.0, 122.5, 122.1, 56.1, 52.7, 24.8, 24.5, 14.4, 13.7. IR (KBr; cm−1): 3023(w), 2965(w), 2361(s), 1677(s), 1651(m), 1597(s), 1549(m), 1493(s), 1439(s), 1257(m), 1193(m), 1080(m), 1032(m), 924(m), 891(m), 830(m), 697(vs). Anal. calcd for C54H43N2Cl (755.39): C, 85.86; H, 5.74; N, 3.71. Found: C, 85.74; H, 5.79; N, 3.68.
1-(2,4-Dibenzhydryl-6-chlorophenylimino)-2-(2,6-diisopropyl-phenylimino)acenaphthylene (L3). Similarly compound L3 was synthezised by the reaction of 2-(2,4-dibenzhydryl-6-chlorophenylimino)acenaphthylenone (1.20 g, 1.92 mmol), 2,6-diisopropylaniline (0.51 g, 2.89 mmol) and p-toluenesulfonic acid (0.2 g), as the yellow powder, 0.35 g (23%). Mp: 208–210 °C. 1H NMR (400 MHz, CDCl3, TMS): δ 7.76 (t, J = 8.6 Hz, 2H), 7.33–7.28 (m, 7H), 7.24–7.11 (m, 12H), 7.00 (d, J = 6.8 Hz, 2H), 6.88 (d, J = 7.2 Hz, 2H), 6.77 (s, 1H), 6.54–6.45 (m, 4H), 6.27 (t, J = 7.4 Hz, 1H), 5.80 (s, 1H), 5.51 (s, 1H), 3.23–3.16 (m, 1H), 3.97–2.91 (m, 1H), 1.33 (d, J = 6.8 Hz, 3H), 1.20 (d, J = 6.8 Hz, 3H), 1.13 (d, J = 6.8 Hz, 3H), 0.86 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3, TMS): δ 163.9, 161.5, 147.3, 143.7, 143.6, 142.7, 141.2, 140.3, 136.2, 135.7, 135.6, 130.5, 129.9, 129.7, 129.5, 129.4, 129.2, 128.9, 128.8, 128.5, 128.2, 127.8, 127.6, 127.5, 126.6, 126.3, 125.6, 124.6, 123.8, 123.4, 123.2, 123.1, 122.2, 56.2, 52.8, 28.7, 28.6, 23.8, 23.7, 23.6, 23.3. IR (KBr; cm−1): 3058(w), 2965(m), 2361(m), 1680(m), 1653(m), 1597(m), 1549(m), 1493(s), 1439(s), 1324(w), 1253(m), 1187(m), 1079(m), 1033(m), 923(m), 890(m), 780(s), 698(vs). Anal. calcd for C56H47N2Cl (783.44): C, 85.85; H, 6.05; N, 3.58. Found: C, 85.53; H, 6.20; N, 3.56.
1-(2,4-Dibenzhydryl-6-chlorophenylimino)-2-(2,4,6-trimethyl-phenylimino)acenaphthylene (L4). Extensively compound L4 was obtained from the reaction of 2-(2,4-dibenzhydryl-6-chlorophenylimino)acenaphthylenone (1.87 g, 3.00 mmol), 2,4,6-trimethylaniline (0.61 g, 4.50 mmol) and p-toluenesulfonic acid (0.2 g), as the yellow powder, 0.40 g (18%). Mp: 140–142 °C. 1H NMR (400 MHz, CDCl3, TMS): δ 7.78 (t, J = 9.6 Hz, 2H), 7.34–7.29 (m, 5H), 7.24–7.12 (m, 10H), 7.01–6.97 (m, 5H), 6.89 (d, J = 7.6 Hz, 2H), 6.77 (s, 1H), 6.66 (d, J = 7.2 Hz, 1H), 6.53–6.47 (m, 3H), 6.27 (t, J = 7.2 Hz, 1H), 5.80 (s, 1H), 5.51 (s, 1H), 2.39 (s, 3H), 2.24 (s, 3H), 2.05 (s, 3H). 13C NMR (100 MHz, CDCl3, TMS): δ 163.7, 161.2, 146.7, 145.4, 143.6, 143.5, 142.5, 141.1, 140.4, 140.2, 136.1, 133.0, 130.3, 129.6, 129.2, 129.1, 128.9, 127.9, 127.3, 126.2, 125.4, 124.6, 124.6, 123.0, 122.2, 122.1, 56.1, 53.4, 52.7, 31.6, 22.7, 20.9, 18.1, 17.1, 14.1. IR (KBr; cm−1): 2959(w), 2923(w), 2361(s), 1669(m), 1644(m), 1597(m), 1550(w), 1443(s), 1233(m), 1075(m), 1033(m), 922(m), 889(w), 783(s), 698(vs). Anal. calcd for C53H41N2Cl (741.36): C, 85.86; H, 5.57; N, 3.78. Found: C, 85.40; H, 5.88; N, 3.63.
1-(2,4-Dibenzhydryl-6-chlorophenylimino)-2-(2,6-diethyl-4-methylphenylimino)acenaphthylene (L5). The compound L5 was similarly prepared by the reaction of 2-(2,4-dibenzhydryl-6-chlorophenylimino)acenaphthylenone (1.25 g, 2.00 mmol), 2,6-diethyl-4-methylaniline (0.49 g, 3.00 mmol) and p-tolunenesulfonic acid (0.2 g), as the orange powder in 0.37 g (24%). Mp: 134–136 °C. 1H NMR (400 MHz, CDCl3, TMS): δ 7.76 (t, J = 9.0 Hz, 2H), 7.31 (t, J = 7.2 Hz, 5H), 7.24–7.11 (m, 11H), 7.04–7.00 (m, 4H), 6.89 (d, J = 7.6 Hz, 2H), 6.77 (s, 1H), 6.64 (d, J = 6.8 Hz, 1H), 6.52–6.47 (m, 3H), 6.28 (t, J = 7.2 Hz, 1H), 5.80 (s, 1H), 5.51 (s, 1H), 2.73–2.63 (m, 1H), 2.59–2.46 (m, 2H), 2.43 (s, 3H), 2.37–2.25 (m, 1H), 2.23 (t, J = 7.4 Hz, 3H), 1.06 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3, TMS): δ 163.8, 161.3, 145.8, 145.4, 143.6, 143.5, 142.6, 141.1, 140.4, 140.1, 136.1, 133.2, 130.6, 130.3, 129.8, 129.6, 129.3, 129.2, 128.7, 128.5, 128.4, 128.0, 127.7, 127.6, 127.3, 127.2, 127.0, 126.5, 126.2, 125.4, 123.0, 122.6, 122.1, 56.1, 52.7, 24.8, 24.5, 21.2, 14.6, 13.8. IR (KBr; cm−1): 2966(w), 2361(m), 1678(m), 1651(m), 1598(m), 1549(s), 1493(s), 1443(s), 1257(m), 1153(w), 1079(m), 1031(m), 922(m), 891(m), 779(s), 738(s), 699(vs). Anal. calcd for C55H45N2Cl (769.41): C, 85.86; H, 5.90; N, 3.64. Found: 85.75; H, 5.85; N, 3.65.
1-(2,4-Dibenzhydryl-6-chlorophenylimino)-2-(2,6-dimethyl phenylimino)acenaphthylylnickel dibromide (Ni1). Compound L1 (0.22 g, 0.30 mmol) and (DME)NiBr2 (0.083 g, 0.27 mmol) were mixed in 10 mL dichloromethane, and stirred at room temperature for 24 h. The solution was concentrated and then 20 mL diethyl ether was added to precipitate resulting complex, which was further washed by diethyl ether and given red nickel complex Ni1, 0.22 g (86%). IR (KBr; cm−1): 2974(w), 2865(w), 2361(s), 1652(m), 1625(s), 1581(s), 1491(m), 1442(s), 1295(s), 1193(w), 1138(w), 1105(s), 1033(m), 920(w), 890(w), 832(m), 776(s), 697(vs). Anal. calcd for C52H39N2ClBr2Ni (945.83): C, 66.03; H, 4.16; N, 2.96. Found: C, 65.88; H, 4.33; N, 3.05.
1-(2,4-Dibenzhydryl-6-chlorophenylimino)-2-(2,6-diethyl phenylimino)acenaphethylylnickel dibromide (Ni2). Using compound L2 instead of L1 in the same procedure in synthesizing Ni1, complex Ni2 was obtained as the brown powder in 0.10 g (88%). IR (KBr; cm−1): 2972(m), 2931(w), 2869(w), 2361(m), 1654(m), 1626(s), 1586(s), 1491(m), 1446(s), 1291(m), 1261(m), 1183(m), 1109(s), 1075(m), 1029(m), 769(s), 741(s), 702(vs). Anal. calcd for C54H43N2ClBr2Ni (973.89): C, 66.60; H, 4.45; N, 2.88. Found: C, 66.22; H, 4.79; N, 2.79.
1-(2,4-Dibenzhydryl-6-chlorophenylimino)-2-(2,6-diisopropyl phenylimino)acenaphthylylnickel dibromide (Ni3). Similarly the complex Ni3 was isolated as the red powder in 0.13 g (96%). IR (KBr; cm−1): 2970(m), 2871(w), 2362(s), 1652(m), 1624(m), 1583(m), 1560(w), 1491(m), 1443(s), 1415(m), 1383(m), 1295(m), 1108(s), 1075(m), 1052(m), 919(w), 773(m), 698(vs). Anal. calcd for C56H47N2ClBr2Ni (1001.94): C, 67.13; H, 4.73; N, 2.80. Found: C, 66.76; H, 4.75; N, 2.78.
1-(2,4-Dibenzhydryl-6-chlorophenylimino)-2-(2,4,6-trimethyl phenylimino)acenaphthylylnickel dibromide (Ni4). Extensively complex Ni4 was obtained as the brown powder in 0.12 g (69%). IR (KBr; cm−1): 2977(m), 2362(s), 1650(m), 1624(s), 1593(s), 1494(s), 1442(s), 1295(m), 1231(m), 1156(w), 1075(m), 1032(m), 919(m), 772(s), 697(vs). Anal. calcd for C53H41N2ClBr2Ni (959.86): C, 66.32; H, 4.31; N, 2.92. Found: C, 66.07; H, 4.39; N, 2.92.
1-(2,4-Dibenzhydryl-6-chlorophenylimino)-2-(2,6-diethyl-4-methylphenylimino)acenaphethylylnickel dibromide (Ni5). Extensively complex Ni5 was obtained as the red powder in 0.15 g (84%). IR (KBr; cm−1): 2964(m), 2929(m), 2361(s), 1656(m), 1625(s), 1585(s), 1560(m), 1493(s), 1446(s), 1414(s), 1292(m), 1075(m), 1030(s), 957(w), 923(w), 858(m), 827(m), 773(s), 696(vs). Anal. calcd for C55H45N2ClBr2Ni (987.91): C, 66.87; H, 4.59; N, 2.84. Found: C, 66.43; H, 4.52; N, 2.85.
1-(2,4-Dibenzhydryl-6-chlorophenylimino)-2-(2,6-dimethyl phenylimino)acenaphthylylnickel dichloride (Ni6). Employing the same procedure for Ni1, using the equimolar NiCl2·6H2O instead of its analog (DME)NiBr2, complex Ni6 was obtained as the yellow powder in 0.11 g (74%). IR (KBr; cm−1): 3057(w), 3025(w), 1661(w), 1628(m), 1588(s), 1494(s), 1444(s), 1291(m), 1262(w), 1191(w), 1080(m), 1031(m), 921(w), 888(w), 831(m), 773(s), 739(s), 697(vs). Anal. calcd for C52H39N2Cl3Ni (856.93): C, 72.88; H, 4.59; N, 3.27. Found: C, 72.72; H, 4.35; N, 3.55.
1-(2,4-Dibenzhydryl-6-chlorophenylimino)-2-(2,6-diethyl phenylimino)acenaphthylylnickel dichloride (Ni7). Using the similar synthetic procedure of complex Ni6, but compound L2 was used instead of L1, complex Ni7 was isolated as the yellow powder in 0.11 g (74%). IR (KBr; cm−1): 3058(w), 3026(w), 2964(w), 1665(m), 1633(m), 1588(s), 1559(w), 1494(m), 1445(s), 1415(m), 1291(m), 1264(m), 1186(m), 1079(m), 1033(m), 829(m), 775(s), 743(s), 698(vs). Anal. calcd for C54H43N2Cl3Ni (884.99): C, 73.29; H, 4.90; N, 3.17. Found: C, 73.07; H, 5.00; N, 2.89.
1-(2,4-Dibenzhydryl-6-chlorophenylimino)-2-(2,6-diisopropyl phenylimino)acenaphthylylnickel dichloride (Ni8). Similarly complex Ni8 was obtained as the yellow powder in 0.07 g (43%). IR (KBr; cm−1): 3058(w), 3026(w), 2967(m), 1660(m), 1624(m), 1591(s), 1491(m), 1445(s), 1416(w), 1291(m), 1259(m), 1182(m), 1080(m), 1033(s), 831(m), 779(s), 744(s), 702(vs). Anal. calcd for C56H47N2Cl3Ni (913.04): C, 73.67; H, 5.19; N, 3.07. Found: C, 73.27; H, 5.14; N, 2.79.
1-(2,4-Dibenzhydryl-6-chlorophenylimino)-2-(2,4,6-trimethyl phenylimino)acenaphthylylnickel dichloride (Ni9). Extensively complex Ni9 was isolated as the yellow powder in 0.08 g (71%). IR (KBr; cm−1): 3058(w), 3025(w), 2972(s), 1661(m), 1630(m), 1591(s), 1558(w), 1494(m), 1444(s), 1416(m), 1290(m), 1155(m), 1113(m), 1033(m), 832(m), 774(s), 737(s), 699(vs). Anal. calcd for C53H41N2Cl3Ni (870.96): C, 73.09; H, 4.74; N, 3.22. Found: C, 72.76; H, 4.75; N, 2.88.
1-(2,4-Dibenzhydryl-6-chlorophenylimino)-2-(2,6-diethyl-4-methylphenylimino)acenaphthylylnickel dichloride (Ni10). Extensively complex Ni10 was obtained as the yellow powder in 0.08 g (49%). IR (KBr; cm−1): 3061(w), 3025(w), 2971(m), 1666(m), 1634(m), 1594(s), 1492(m), 1447(s), 1414(m), 1291(m), 1259(m), 1113(m), 1077(m), 1034(s), 773(s), 741(s), 702(vs). Anal. calcd for C55H45N2Cl3Ni (899.01): C, 73.48; H, 5.05; N, 3.12. Found: C, 73.01; H, 5.06; N, 3.19.
X-ray crystallographic study
Single crystals of the nickel complexes Ni2 and Ni5 were obtained by slow diffusion of diethyl ether into dichloromethane solution at room temperature. Single-crystal X-ray diffraction studies for them were carried out on a Rigaku Saturn 724+ CCD with graphite-monochromatic Mo-Kα radiation (λ = 0.71073 Å) at 173(2) K, the cell parameters were obtained by global refinement of the positions of all collected reflections. Intensities were corrected for Lorentz and polarization effects and empirical absorption. The structure was solved by direct methods and refined by full-matrix least squares on F2. All hydrogen atoms were placed in calculated positions. Structure solution and refinement were performed by using the SHELXL-97 package.16 Details of the crystal data and structure refinements for Ni2 and Ni5 are shown in Table 4.
Table 4 Crystal data and structure refinements for Ni2 and Ni5
| |
Ni2 |
Ni5 |
| Empirical formula |
C54H43N2ClBr2Ni·CH2Cl2 |
C55H45N2ClBr2Ni |
| Formula weight |
1058.81 |
987.91 |
| Temperature/K |
291(2) |
173(2) |
| Wavelength/Å |
0.71073 |
0.71073 |
| Crystal system |
Triclinic |
Orthorhombic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P2(1)2(1)2(1) |
| a/Å |
12.0590(13) |
16.470(3) |
| b/Å |
15.8340(14) |
16.715(3) |
| c/Å |
26.7560(15) |
16.782(3) |
| Alpha/° |
88.590(2) |
90 |
| Beta/° |
83.230(3) |
90 |
| Gamma/° |
72.860(2) |
90 |
| Volume/Å3 |
4847.6(7) |
4620.0(16) |
| Z |
4 |
4 |
| Dcalcd/(g cm−3) |
1.451 |
1.420 |
| μ/mm−1 |
2.253 |
2.247 |
| F(000) |
2152 |
2016 |
| Crystal size/mm |
0.37 × 0.14 × 0.08 |
0.44 × 0.43 × 0.26 |
| θ range (°) |
1.35–27.00 |
1.74–27.48 |
| Limiting indices |
−15 ≤ h ≤ 15 |
−21 ≤ h ≤ 21 |
| −20 ≤ k ≤ 20 |
−21 ≤ k ≤ 21 |
| −34 ≤ l ≤ 34 |
−21 ≤ l ≤ 20 |
| No. of rflns collected |
66187 |
31595 |
| No. unique rflns |
21117 |
10563 |
| R(int) |
0.0128 |
0.0765 |
| No. of params |
1139 |
553 |
| Completeness to θ |
99.7% |
99.7% |
| Goodness of fit on F2 |
1.057 |
1.092 |
| Final R indices [I > 2∑(I)] |
R1 = 0.0527 |
R1 = 0.0526 |
| wR2 = 0.0967 |
wR2 = 0.1146 |
| R indices (all data) |
R1 = 0.0757 |
R1 = 0.0574 |
| wR2 = 0.1001 |
wR2 = 0.1174 |
| Largest diff. peak, and hole/(e Å−3) |
0.679 and −0.473 |
0.475 and −0.751 |
Ethylene polymerization
Ethylene polymerization at 1 atm ethylene pressure. The polymerization at 1 atm ethylene pressure was carried out in Schlenk techniques. The complex Ni1 was added, and the require amount of co-catalyst (EASC) was added by a syringe, the solvent is toluene. Then the solution was stirred with 1 atm ethylene atmosphere at 50 °C. After 30 min, the solution was quenched with 10% hydrochloric acid in ethanol. The polymer was washed with ethanol, then dried in vacuum at 60 °C and weighed.
Ethylene polymerization at 10/5 atm ethylene pressure. The polymerization at high ethylene pressure was carried out in stainless steel autoclave (0.25 L) equipped with an ethylene pressure control system, a mechanical stirrer and a temperature controller. At the required reaction temperature, 30 mL toluene (freshly distilled) was injected into the autoclave, then 50 mL toluene which dissolved the complex (2.0 μmol), the require amount of co-catalyst (MAO, MMAO, AlEtCl2,EASC), the another 20 mL toluene were injected by syringe successively. The autoclave was immediately pressurized to high ethylene pressure and the stir started. After the required reaction time, the ethylene was released; the polymer was washed by ethanol, and then dried in vacuum at 60 °C and weighed.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 51373176, 21374123, and U1362204).
Notes and references
-
(a) L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef CAS;
(b) C. M. Killian, D. J. Tempel, L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 11664–11665 CrossRef CAS.
-
(a) C. Bianchini, G. Giambastiani, L. Luconi and A. Meli, Coord. Chem. Rev., 2010, 254, 431–455 CrossRef CAS;
(b) D. H. Camacho and Z. Guan, Chem. Commun., 2010, 46, 7879–7893 RSC;
(c) R. Gao, W.-H. Sun and C. Redshaw, Catal. Sci. Technol., 2013, 3, 1172–1179 RSC;
(d) S. Wang, W.-H. Sun and C. Redshaw, J. Organomet. Chem., 2014, 751, 717–741 CrossRef CAS;
(e) Z. Flisak and W.-H. Sun, ACS Catal., 2015, 5, 4713–4724 CrossRef CAS.
-
(a) S. A. Svejda and M. Brookhart, Organometallics, 1999, 18, 65–74 CrossRef CAS;
(b) D. P. Gates, S. A. Svejda, E. Oñate, C. M. Killian, L. K. Johnson, P. S. White and M. Brookhart, Macromolecules, 2000, 33, 2320–2334 CrossRef CAS;
(c) D. H. Camacho, E. V. Salo, J. W. Ziller and Z. Guan, Angew. Chem., Int. Ed., 2004, 43, 1821–1825 CrossRef CAS PubMed;
(d) C. S. Popency, C. M. Levins and Z. Guan, Organometallics, 2011, 30, 2432–2452 CrossRef.
- M. Schmid, R. Eberhardt, M. Klinga, M. Leskelä and B. Rieger, Organometallics, 2001, 20, 2321–2330 CrossRef CAS.
-
(a) D. Jia, W. Zhang, W. Liu, L. Wang, C. Redshaw and W.-H. Sun, Catal. Sci. Technol., 2013, 3, 2737–2745 RSC;
(b) Q. Liu, W. Zhang, D. Jia, X. Hao, C. Redshaw and W.-H. Sun, Appl. Catal., A, 2014, 475, 195–202 CrossRef CAS;
(c) J. L. Rhinehart, N. E. Mitchell and B. K. Long, ACS Catal., 2014, 4, 2501–2504 CrossRef CAS.
- H. Liu, W. Zhao, X. Hao, C. Redshaw, W. Huang and W.-H. Sun, Organometallics, 2011, 30, 2418–2424 CrossRef CAS.
- S. Kong, C.-Y. Guo, W. Yang, L. Wang, W.-H. Sun and R. Glaser, J. Organomet. Chem., 2013, 725, 37–45 CrossRef CAS.
- L. Fan, S. Du, C.-Y. Guo, X. Hao and W.-H. Sun, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1369–1378 CrossRef CAS.
-
(a) H. Liu, W. Zhao, J. Yu, W. Yang, X. Hao, C. Redshaw, L. Chen and W.-H. Sun, Catal. Sci. Technol., 2012, 2, 415–422 RSC;
(b) S. Du, S. Kong, Q. Shi, J. Mao, C. Guo, J. Yi, T. Liang and W.-H. Sun, Organometallics, 2015, 34, 582–590 CrossRef CAS;
(c) S. Du, Q. Xing, Z. Flisak, E. Yue, Y. Sun and W.-H. Sun, Dalton Trans., 2015, 44, 12282–12291 RSC.
-
(a) D. Guo, L. Han, T. Zhang, W.-H. Sun, T. Li and X. Yang, Macromol. Theory Simul., 2002, 11, 1006–1012 CrossRef CAS;
(b) T. Zhang, D. Guo, S. Jie, W.-H. Sun, T. Li and X. Yang, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4765–4774 CrossRef CAS;
(c) W. Yang, J. Yi and W.-H. Sun, Macromol. Chem. Phys., 2015, 216, 1125–1133 CrossRef CAS.
-
(a) E. Yue, L. Zhang, Q. Xing, X.-P. Cao, X. Hao, C. Redshaw and W.-H. Sun, Dalton Trans., 2014, 43, 423–431 RSC;
(b) E. Yue, Q. Xing, L. Zhang, Q. Shi, X.-P. Cao, L. Wang, C. Redshaw and W.-H. Sun, Dalton Trans., 2014, 43, 3339–3346 RSC;
(c) F. Huang, Z. Sun, S. Du, E. Yue, J. Ba, X. Hu, T. Liang, G. B. Galland and W.-H. Sun, Dalton Trans., 2015, 44, 14281–14292 RSC.
- C. Wen, S. Yuan, Q. Shi, E. Yue, D. Liu and W.-H. Sun, Organometallics, 2014, 33, 7223–7231 CrossRef CAS.
-
(a) D. J. Tempel, L. K. Johnson, R. L. Huff, P. S. White and M. Brookhart, J. Am. Chem. Soc., 2000, 122, 6686–6700 CrossRef CAS;
(b) R. Chen and S. F. Mapolie, J. Mol. Catal. A: Chem., 2003, 193, 33–40 CrossRef CAS;
(c) J. Lai, W. Zhao, W. Yang, C. Redshaw, T. Liang, Y. Liu and W.-H. Sun, Polym. Chem., 2012, 3, 787–793 RSC.
- G. B. Galland, R. F. de Souza, R. S. Mauler and F. F. Nunes, Macromolecules, 1999, 32, 1620–1625 CrossRef CAS.
-
(a) Q. Yang, M. D. Jensen and M. P. McDaniel, Macromolecules, 2010, 43, 8836–8852 CrossRef CAS;
(b) V. Karimkhani, F. A.- Taromi, S. Pourmahdian and F. J. Stadler, Polym. Chem., 2013, 4, 3774–3790 RSC.
- G. M. Sheldrick, SHELXTL-97, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1422645 and 1422646 contain the supplementary crystallographic data for Ni2 and Ni5. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra18257c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.