
Self-microemulsifying drug delivery system of curcumin with enhanced solubility and bioavailability using a new semi-synthetic bicephalous heterolipid: in vitro and in vivo evaluation†
Dinesh M. Dhumala,
Priya R. Kotharia,
Rahul S. Kalhapureb and
Krishnacharya G. Akamanchi*a
aDepartment of Pharmaceutical Sciences and Technology, Institute of Chemical Technology, Matunga (E), Mumbai 400019, India. E-mail: kgap@rediffmail.com; Fax: +91-22-33611020; Tel: +91-22-33612214
bDiscipline of Pharmaceutical Sciences, School of Health Sciences, University of KwaZulu-Natal, Private Bag X54001, Durban 4000, KwaZulu-Natal, South Africa
First published on 13th October 2015
Abstract
Despite the various therapeutic actions displayed by curcumin, its poor aqueous solubility and bioavailability restricts its biological applications. This problem can be addressed by its efficient loading into a nano formulation. The present paper reports the utilization of a new semi-synthetic oleic acid derived bicephalous heterolipid, E1E, as an oil phase in the formulation development of a self-microemulsifying drug delivery system (SMEDDS) of curcumin to enhance its solubility and bioavailability. The solubility of curcumin in E1E was found to be 14 and 2.6-fold greater than oleic acid and ethyl oleate respectively. The SMEDDS developed from E1E (Curcumin-E1E_SMEDDS5e) had high curcumin loading efficiency of 70.52 ± 2.46 mg g−1, and was able to form spontaneous microemulsion on addition to aqueous phase with mean globule diameter of 22.39 ± 0.2 nm and polydispersity index of 0.243 ± 0.010. In vivo oral bioavailability studies in male Wistar rats revealed that the maximum serum concentration (Cmax) and time taken to reach maximum serum concentration (Tmax) were 4.921 ± 0.42 μg mL−1 and 60 min respectively. The absorption of curcumin increased 26-folds via its delivery through Curcumin-E1E_SMEDDS5e. The cytostatic and total growth inhibition concentrations of Curcumin-E1E_SMEDDS5e against HeLa cell line were almost comparable to doxorubicin (GI50 and TGI < 10 μg mL−1) whereas lethal concentration (LC50) was 71.8 μg mL−1. Further, the minimum inhibitory concentrations against E. coli and S. aureus were 48.62 and 97.65 μg mL−1 respectively. These results suggested potent anticancer, antibacterial activity and non-cytotoxicity of developed Curcumin-E1E_SMEDDS5e. The heterolipid, E1E, proved to be an efficient solubility and bioavailability enhancer for curcumin for formulation development of its SMEDDS.
Introduction
Curcumin is a naturally occurring phytochemical found in turmeric and is commonly used as a food preservative, spice, coloring and flavoring agent.1 Chemically it is [(E,E)-1,7-bis(4-hydroxy-3-methoxy-phenyl)-1,6-heptadiene-3,5-ione and has emerged as a powerful “nutraceutical” which regresses diseases safely and inexpensively by interacting with multiple targets.1,2 It is a very safe medication as indicated by the fact that up to 8 g per day for 18 months was non-toxic to humans.2 It exhibits various therapeutic actions such as anti-inflammatory, anticancer, antibacterial, antirheumatic, antimalarial, antioxidant3–6 and pharmacological properties such as cardioprotective, nephroprotective, neuroprotective and hepatoprotective.7 Despite several uses in pharamacotherapeutics, curcumin's applications are severely restricted due to its short half-life, very low aqueous solubility, poor absorption from the gut and extremely low bioavailability.8–10 A maximum serum concentration of curcumin detected in rats after oral dose of 2 g kg−1 was found to be 1.35 ± 0.23 μg mL−1, whereas same dose in humans resulted in either undetectable or extremely low serum levels after 1 h.11 Curcumin, a phyto-derived active constituent can be a promising clinical drug for the treatment of cancer and bacterial infections in current drug resistance scenario if the problems associated with its delivery are addressed.Oral route is the most preferred route of drug administration and will continue to be as it provides large surface area with changing environments for absorption, encompassed with different systems of blood circulation which provide opportunities to bypass liver to overcome first pass effects. Suitability of a drug molecule for oral administration is measured on the basis of several parameters including solubility and stability in gastrointestinal fluid, bio-stability towards digestive enzymes, and biodegradation due to intestinal micro flora. Unfortunately, very few drug molecules fulfill these requirements and others pose problem for oral administration.12 It is anticipated that up to 70% of newly identified bioactive compounds with potential to emerge as drugs could fall under biopharmaceutics classification system II (low solubility and high permeability) or IV (low solubility and permeability).13 Curcumin's poor solubility and bioavailability problems can be solved effectively via formulating its nano drug delivery systems. Therefore, various nanoparticle based systems such as polymeric nanoparticles,14 liposomes,15 nanogels,16 solid lipid nanoparticles,17 polymer conjugate,18 and curcumin–phospholipid complex19 have been reported to enhance its oral bioavailability. Though these deliveries have shown some advantages along with increased oral bioavailability, the utilization of techniques such as complexation, conjugation, and tedious manufacturing processes restricts their scale up and commercialization. Therefore, there is a need to develop more convenient and cost effective nano delivery systems of curcumin.
Lipid based nano drug delivery systems is an attractive strategy due to its advantages over other systems and recently formulation scientists have developed and reported various lipid based curcumin nano formulations.6,20–23 Amongst various lipid based drug delivery systems self-microemulsifying drug delivery systems (SMEDDS) is an attractive approach to improve solubility and oral bioavailability of poorly soluble drugs.24 SMEDDS, an isotropic mixture of oil and surfactants, forms an emulsion with mild agitation in the gastrointestinal fluid and is an anticipated technology to increase the water solubility of poorly water-soluble drugs due to their large solubilization capacity and small droplet size. These properties of SMEDDS allow enhanced permeation and bioavailability of the formulated drug across the gastrointestinal membrane.25 In recent years, curcumin SMEDDSs for various purposes such as treatment of ulcerative colitis,26 improvement of dissolution, solubility and oral bioavailability,27–30 and colon targeting for cancer treatment31 have been prepared and evaluated. These are very few reports on curcumin delivery via SMEDDS though the use of SMEDDS is regarded as a smart approach because of its transparency, thermodynamic stability, simplicity of scale up and manufacture.32–34 One of the reasons could be a limited number of available effective lipids (solubility and bioavailability enhancers) for the development of curcumin SMEDDS as most of the papers report the use of Capryol™ 90,26,29–31 as an oily phase with few exceptions where Labrafac PG,30 isopropyl myristate, soybean oil and aethylisoleas28 are reported. Therefore, there is a need for newer and tailor made semi-synthetic lipids with structural diversity for the formulation development of curcumin SMEDDS in order to enhance its solubility, permeability and bioavailability.
Our research group is engaged in the development of new lipids particularly heterolipids with amphiphilic properties having multiple head groups with a scope for structural modifications. One such heterolipid synthesized by attaching dendritic head group to oleic acid moiety and has been developed as an excipient is (Z)-di-tert-butyl 3,3′-((3-(oleoyloxy)propyl)azanediyl)dipropanoate (designated as heterolipid E1E). Its structural features are ester linkage between oleoyl moiety and head group, a protonable tertiary amine, and tertiary ester group at its periphery (Fig. 1). This bicephalous heterolipid E1E has been evaluated for its physico-chemical and biological properties, was found to be non-toxic to mammalian cells paving the way towards its biological applications. Its pharmaceutical uses as a new excipient have been demonstrated by employing it as an oily phase in the formulation of furosemide SMEDDS35 and as a transdermal permeation enhancer for diclofenac sodium.36
 | ||
| Fig. 1 Chemical structure of heterolipid E1E. | ||
The study reported in this paper was focused on the utilization of E1E in the formulation development of curcumin SMEDDS with increased solubility and enhanced oral bioavailability. Our hypothesis was E1E could act as (a) a good solubilizer by ionic interaction, hydrogen bonding and hydrophobic drug–lipid interactions in between acidic phenol groups in curcumin and protonable basic nitrogen in the structure of E1E and (b) a bioavailability enhancer due to its permeation enhancement property. The promising results such as increased solubility, enhanced oral bioavailability, anticancer and antibacterial activity obtained through formulation development of curcumin SMEDDS containing E1E as an oily phase are discussed here.
Materials and methods
Materials
Curcumin was obtained as a gift sample from Konark Herbals Ltd., India. Oleic acid (technical grade, 90%) was purchased from Sigma, USA. Solutol HS 15, Cremophore RH40, Cremophore EL and Labrasol were kindly provided as gift samples by BASF, India. Transcutol HP was a generous gift sample from Gattefosse, India. Ethyl oleate, Tween 20, Tween 80, PEG-400, propylene glycol, isopropyl alcohol and ethanol were purchased from S D Fine-Chem Ltd, India. All the solvents used were of HPLC grade and obtained from Merck. Water was obtained from the Milli-Q water purification system of Millipore Corporation (Bedford, USA).Solubility studies
Solubility in oils was determined by adding an excess amount of curcumin in 1 g of ethyl oleate, oleic acid and synthesized heterolipid E1E (ESI†). Solubility in 10% w/w aqueous solutions of surfactants (Cremophor RH40, Cremophor EL, Labrasol, Tween 80, Tween 20, Solutol HS15), and co-surfactants (ethanol, isopropyl alcohol, propylene glycol, Transcutol HP, PEG-400) was determined separately by adding excess curcumin to 1 g of selected surfactant and co-surfactant solution. The mixtures were vortexed for 10 min and kept at 25 ± 2 °C in an isothermal shaker for 48 h to attain equilibrium. The equilibrated samples were removed from the shaker and centrifuged at 5000 rpm for 15 min. The supernatant was collected and filtered through a 0.45 μm membrane filter (Pall Life Sciences) and the filtrate was analyzed for curcumin by few modifications in a high performance liquid chromatographic (HPLC) method reported in the literature.37 A HPLC system (Jasco, Japan) consisting of a Jasco-PU-1580 pump, a Rheodyne injection system with a loop of 20 μL was used. The separation was performed on a LiChrospher®100 RP-C18-column, 5 μm (250 mm × 4.6 mm i.d.). An isocratic elution was performed using mobile phase consisting of 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 v/v of acetonitrile: 2% v/v acetic acid solution in Milli-Q water, flow rate was 1 mL min−1 and samples were analyzed at wavelength of 425 nm. The assay method was validated and the calibration curve was linear (r2 = 0.999) within the concentration range of 0.1–20 μg mL−1.
:40 v/v of acetonitrile: 2% v/v acetic acid solution in Milli-Q water, flow rate was 1 mL min−1 and samples were analyzed at wavelength of 425 nm. The assay method was validated and the calibration curve was linear (r2 = 0.999) within the concentration range of 0.1–20 μg mL−1.
Screening of surfactants and co-surfactants
To evaluate the capability of oil and surfactants to form an emulsion spontaneously, homogenous mixtures consisting of heterolipid E1E with different surfactants in the ratio of 3:1 (w/w) were vortexed and heated at 45–60 °C. Each of isotropic mixtures (50 mg) were diluted with Milli-Q water (50 mL) to obtain an emulsion. The emulsions were allowed to stand for 2 h and their transmittance was measured at λmax 632.8 nm. Spectra were recorded on UV-1650 PC, UV-visible spectrophotometer (Shimadzu, Japan) using Milli-Q water as blank.38
For the screening of co-surfactants, different surfactant:co-surfactant mixtures (Smix) consisting of 0.2 g of screened surfactant and 0.1 g of co-surfactants were prepared. To these Smix about 0.3 g of heterolipid E1E was added and these mixtures were homogenized with the aid of gentle heat (45–60 °C). The resultant isotropic mixtures were accurately weighed (50 mg) and diluted to 50 mL with Milli-Q water to yield fine emulsion. The emulsions so formed were analyzed spectrophotometrically in a similar manner as that of surfactants.
Construction of pseudo-ternary phase diagrams
Heterolipid E1E was selected as an oil phase based on the preliminary screening experiments whereas Solutol HS 15 was used as a surfactant and Transcutol HP as a co-surfactant. Milli-Q water was used as an aqueous phase for construction of phase diagrams. At Km (surfactant:co-surfactant ratio) values of 1, 1.5 and 3, mixtures of oil, surfactant and co-surfactant were prepared by vortexing wherein the ratio of oil to surfactant/co-surfactant blend was varied from 9:1 to 1:9. Each mixture was then slowly titrated with aliquots of Milli-Q water and stirred at room temperature to attain equilibrium. The mixture was visually examined for transparency. After equilibrium was reached, the mixtures were further titrated with aliquots of Milli-Q water until they showed presence of turbidity. The clear and isotropic samples were deemed to be within the microemulsion region. Several compositions were selectively scrutinized on the basis of their ease to self-emulsify under dilution. The dispersions, having globule size below 100 nm were considered desirable. The micro-emulsions were observed for 48 h and those showing phase separation were rejected. Based on the results, appropriate percentages of oil, surfactant and co-surfactant were selected, correlated in the phase diagram and were used for preparation of SMEDDS containing curcumin.
Optimization of formula
Based on the pseudo-ternary phase diagram studies, five SMEDDS (Curc-E1E_SMEDDS1 to Curc-E1E_SMEDDS5) at desired component ratios were selected and tested for thermodynamic stability, dispersion quality and drug loading efficiency.39,40 Optimized composition was selected based on micro-emulsification area, fast dispersion, drug loading efficiency and physical stability of formed micro-emulsions.Thermodynamic testing
Heating cooling cycle: The formulations were subjected to six cycles of exposure to 4 °C and 45 °C with storage at each temperature of not less than 48 h. Those formulations, which were stable at these temperatures, were subjected to centrifugation test. Centrifugation test: Centrifugation was performed at 3500 rpm for 30 min. Those formulations that did not show any phase separation were evaluated for freeze thaw stress test. Freeze thaw cycle test: The formulations were stored at −21 °C for 48 h and later stored for another 48 h at +25 °C and then observed for any signs of phase separation. Those formulations, which passed these thermodynamic tests, were further exposed to the dispersibility test for assessing efficiency of self-emulsification.Dispersibility test
Efficiency of self-emulsification of SMEDDS was assessed using visual test. Performance of the formulations was visually assessed using the following grading system:39,40Grade A: rapidly forming (within 1 min) emulsion, having a clear or bluish appearance.
Grade B: rapidly forming, slightly less clear emulsion, having a bluish white appearance.
Grade C: fine milky emulsion that formed within 2 min.
Grade D: dull, grayish white emulsion having slightly oily appearance that is slow to emulsify (longer than 2 min).
Drug loading efficiency
Each of the thermodynamically stable and well dispersible blank emulsion was tested for loading efficiency. Curcumin was loaded into 1 g of each blank SMEDDS till the saturation level was reached. The loading in each formulation was determined using HPLC method given under solubility studies. The composition that was thermodynamically stable, well dispersible (Grade A) and having good drug loading capacity was selected. It was critically observed that curcumin loading significantly altered some properties of SMEDDS; hence further optimization of formulation was carried out by determining the effect of drug loading on selected formulation (Curc-E1E_SMEDDS5).Effect of drug loading on Curc-E1E_SMEDDS5
Since thermodynamically stable formulation Curc-E1E_SMEDDS5 exhibited good dispersibility and drug loading, a series of Curc-E1E_SMEDDS5 with variable curcumin loading (viz., 10–70 mg g−1 of SMEEDS) formulations were prepared (Curc-E1E_SMEDDS5a-g) and 50 mg of each formulation was dispersed in 50 mL of Milli-Q water. These dispersed formulations were assessed for 48 h in terms of drug precipitation, mean globule size, polydispersity index (PDI), drug content and % transmission. The most stable and efficient formulation Curc-E1E_SMEDDS5e was selected and evaluated further.Characterization of curcumin-SMEDDS
:25 and mixed by gentle shaking, a drop of it was placed on copper grid and stained in 5% uranyl acetate for 10 min. Any excess of liquid was drawn off with filter paper, grid surface was then air dried at room temperature and examined using TEM at an accelerating voltage of 120 kV.30000 rpm at 0 °C for 15 min, plasma (100 μL) was separated, and diluted by adding acetonitrile to make up to 1 mL to precipitate blood proteins and again centrifuged at 12000 rpm 20 °C for 15 min, and the supernatant was directly injected into the HPLC column (ESI†). Three separate time analysis (n = 3) were performed for each sample. Data from these samples were used to plot curves for curcumin absorption with time. Main pharmacokinetic parameters determined for Curc-E1E_SMEDDS5e and curcumin suspension administered were maximum concentration (Cmax), time to reach maximum concentration (Tmax), and area under the concentration–time curve (AUC0–∞).In vitro anticancer activity. Anticancer activity of Curc-E1E_SMEDDS5e was determined against human cervix cancer cell line (HeLa) by sulforhodamine B (SRB) assay at different curcumin concentrations (10, 20, 40 and 80 μg mL−1) using doxorubicin as a positive control.35,41,42 The cell lines were grown in RPMI 1640 medium containing 10% fetal bovine serum and 2 mM L-glutamine. For present screening experiment, cells were inoculated into 96 well microtiter plates in 100 μL at plating densities depending on the doubling time of individual cell lines. After cell inoculation, the microtiter plates were incubated at 37 °C, 5% CO2, 95% air and 100% relative humidity for 24 h prior to addition of experimental substances. Experimental substances were initially solubilized in water at 100 mg mL−1 and diluted to 1 mg mL−1 using water and stored frozen prior to use. At the time of addition, an aliquot of frozen concentrate (1 mg mL−1) was thawed and diluted to 100 μg mL−1, 200 μg mL−1, 400 μg mL−1 and 800 μg mL−1 with complete medium containing test substance. Aliquots of 10 μL of these different dilutions were added to the appropriate microtiter wells already containing 90 μL of medium, resulting in the required final drug concentrations to 10 μg mL−1, 20 μg mL−1, 40 μg mL−1 and 80 μg mL−1.
After compound addition, plates were incubated at standard conditions for 48 h and assay was terminated by the addition of cold TCA. Cells were fixed in situ by the gentle addition of 50 μL of cold 30% (w/v) TCA (final concentration, 10% TCA) and incubated for 60 minutes at 4 °C. The supernatant was discarded; the plates were washed five times with tap water and air dried. SRB solution (50 μL) at 0.4% (w/v) in 1% acetic acid was added to each of the wells, and plates were incubated for 20 minutes at room temperature. After staining, unbound dye was recovered and the residual dye was removed by washing five times with 1% acetic acid. The plates were air dried, bound stain was subsequently eluted with 10 mM Trizma base, and the absorbance was read on a plate reader at a wavelength of 540 nm with 690 nm reference wavelength.
The percentage growth was calculated at each of the concentration levels using the six absorbance measurements. The dose response parameters were calculated for each test article. Values were calculated for three parameters; concentration of drug causing 50% inhibition of cell growth (GI50), concentration of drug causing total inhibition of cell growth (TGI), and concentration of drug causing 50% cell kill (LC50).
Percentage growth was calculated on a plate-by-plate basis for test wells relative to control wells. Percentage growth was expressed as the ratio of average absorbance of the test well to the average absorbance of the control wells multiplied by 100. Using the six absorbance measurements [time zero (Tz), control growth (C), and test growth in the presence of drug at the four concentration levels (Ti)], the percentage growth was calculated at each of the drug concentration levels. Percentage growth inhibition was calculated as: growth% = Ti/C × 100.
In vitro antimicrobial activity. The minimum inhibitory concentrations (MICs) were determined by following a broth micro-dilution technique (ESI†). The bacterial strains used were E. Coli (ATCC 25922) and S. aureus (ATCC 29213) which are the representative bacteria from Gram negative and Gram positive class respectively. Bacterial isolates were removed from storage, streaked on to Mueller-Hinton Agar (MHA) plates and incubated for 24 h at 35 °C. A working bacterial suspension was prepared by suspending 3–5 isolated colonies in 3 mL of Mueller-Hinton Broth (MHB). The turbidity of this suspension was carefully adjusted photometrically to equal that of a 0.5 McFarland standard. Within 15 minutes of preparation, inoculum suspension was adjusted with MHB so that each well contains approximately 5 × 105 CFU mL−1.
Stability studies. The stability test was performed according to the ICH guidelines (2003) on the topic of Q 1 A (R2): stability testing of new drug substances and products.43 The Curc-E1E_SMEDDS5e were manually filled in hard gelatin capsules size 00 (n = 3). These capsules were stored in air-tight glass containers and protected from light. These capsules were maintained in a stability chamber (Thermo lab, India) under long term study conditions [25 ± 2 °C/60% ± 5% relative humidity (RH)]; intermediate study conditions [30 ± 2 °C/65 ± 5% (RH)], and accelerated study conditions (45 ± 2 °C/75 ± 5% RH). The samples were evaluated at specified time frequency (0, 1, 2 and 3 month) for dispersibility, mean globule size, PDI and drug content.
Results and discussion
Solubility studies
Lipophilic drugs such as curcumin are preferably solubilized in o/w microemulsion. Higher solubility in oil phase helps to minimize the volume of the formulation to deliver the therapeutic dose of the drug in an encapsulated form.44 It was observed that curcumin exhibited greater solubility in heterolipid E1E, a basic lipid compared to oleic acid, an acidic lipid and ethyl oleate, a neutral lipid (Fig. S4, ESI†). The heterolipid E1E could have acted as a good solubilizer by ionic interaction, hydrogen bonding and hydrophobic drug–lipid interactions in between acidic phenol groups in curcumin and a presence of basic tertiary nitrogen in the dendritic head group. These results support the observation that ionic interactions, hydrogen bonding and hydrophobic drug–dendrimer interactions are the mechanisms by which dendrimers exert their solubilizing effect.45 The solubility of curcumin in heterolipid E1E was found to be 0.143% w/w which was 14-fold greater than oleic acid (0.01% w/w) and 2.6-fold greater than ethyl oleate (0.05% w/w). The solubility of curcumin in various surfactants was in the following order: Solutol HS 15 > Tween 20 > Cremophor EL > Labrasol > Cremophor RH > Tween 80 (Fig. S5, ESI†).Among the co-surfactants, PEG 400 (1.035% w/w) and Transcutol HP (0.8112% w/w) showed highest solubility of curcumin (Fig. S6, ESI†). Hence, PEG 400 and Transcutol HP were selected as co-surfactants and were assessed further for their ease of emulsification in order to arrive at final co-surfactant to be used in SMEDDS.
Screening of surfactants and co-surfactants for emulsifying ability
Screening of surfactants and co-surfactants was governed by their emulsification efficiency for the selected oil rather than their ability to solubilize curcumin. Testing of emulsification capacity with the help of transmittance% study revealed that Solutol HS15 was most efficient for emulsifying heterolipid E1E in water (Fig. 2A). The results obtained indicated that structure, bonding interaction and relative length of hydrophobic chains of surfactants had influence on micro-emulsification.46 Therefore, Solutol HS 15 was selected for further study.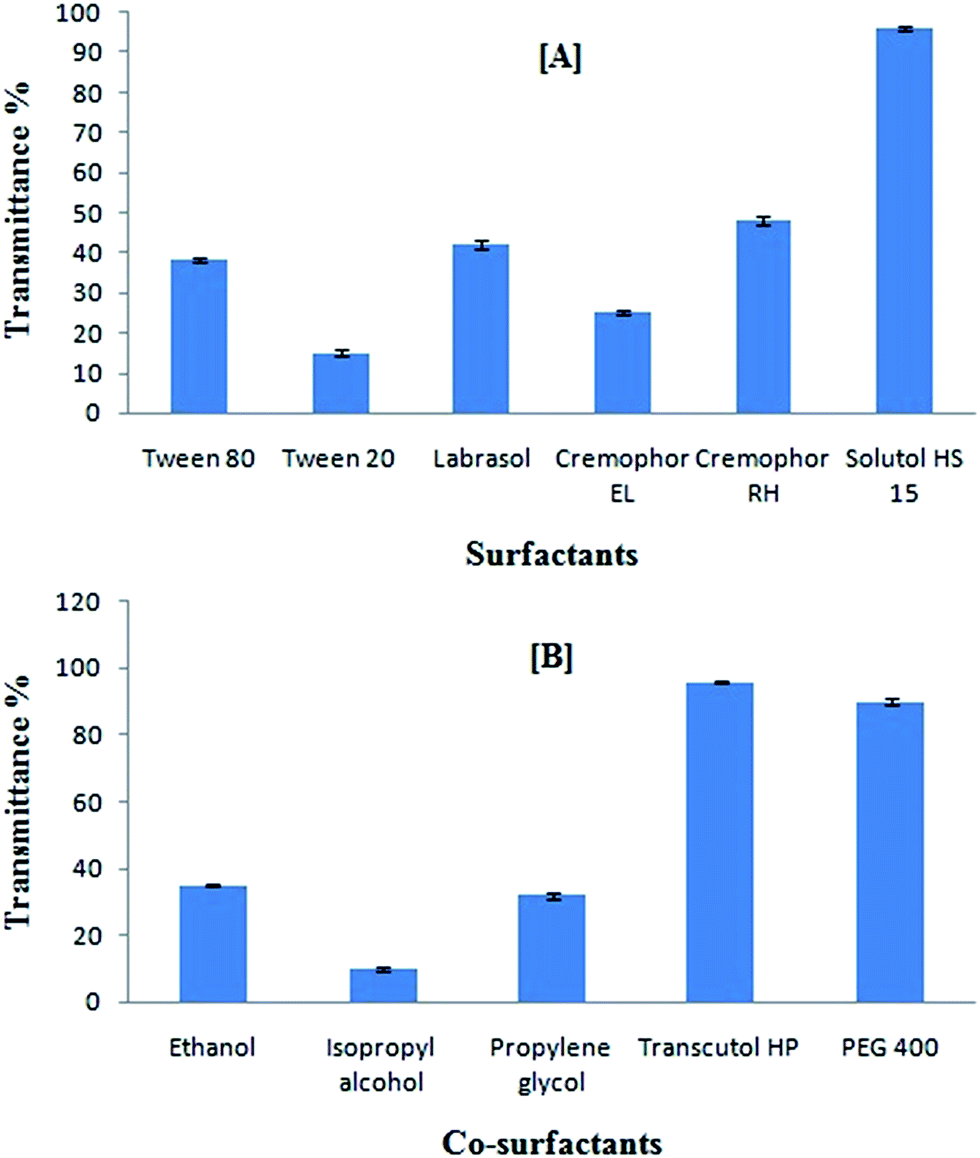 | ||
| Fig. 2 (A) Emulsification efficiency of various surfactants with heterolipid E1E, data expressed as mean ± SD (n = 3); (B) emulsification efficiency of various co-surfactants with heterolipid E1E and surfactant Solutol HS15, data expressed as mean ± SD (n = 3). | ||
The transmittance% study (Fig. 2B) for the combined effect of various surfactants and co-surfactants mixture on heterolipid E1E as an oil phase in emulsion formation revealed that in the presence of Transcutol HP, the spontaneity of microemulsion formation was increased along with the transmittance compared to that achieved in the case of Solutol HS 15 alone. This suggested that Transcutol HP made an interfacial film of sufficient flexibility to take up different curvatures required to form microemulsion. The observed high transmittance% suggested that Transcutol HP was successful in reducing the interfacial tension to a much greater extends than other co-surfactants.
Construction of pseudo-ternary phase diagrams
Pseudo-ternary phase diagrams were constructed to identify microemulsion regions and to optimize the concentration of the selected components viz., heterolipid E1E, Solutol HS15 and Transcutol HP. It can be observed (Fig. 3) that microemulsion region decreased with an increase in concentration of Transcutol HP (i.e. Km value of 1 > 3 > 1.5) but simultaneously it was observed that increasing concentration of Transcutol HP not only helped in decreasing the viscosity but also aided in quick emulsification. Hence, in order to avail the combined benefits of quick emulsification, low viscosity and high drug loading of curcumin, some selected compositions among all three pseudoternary graphs were subjected to thermodynamic, dispersibility and drug loading test.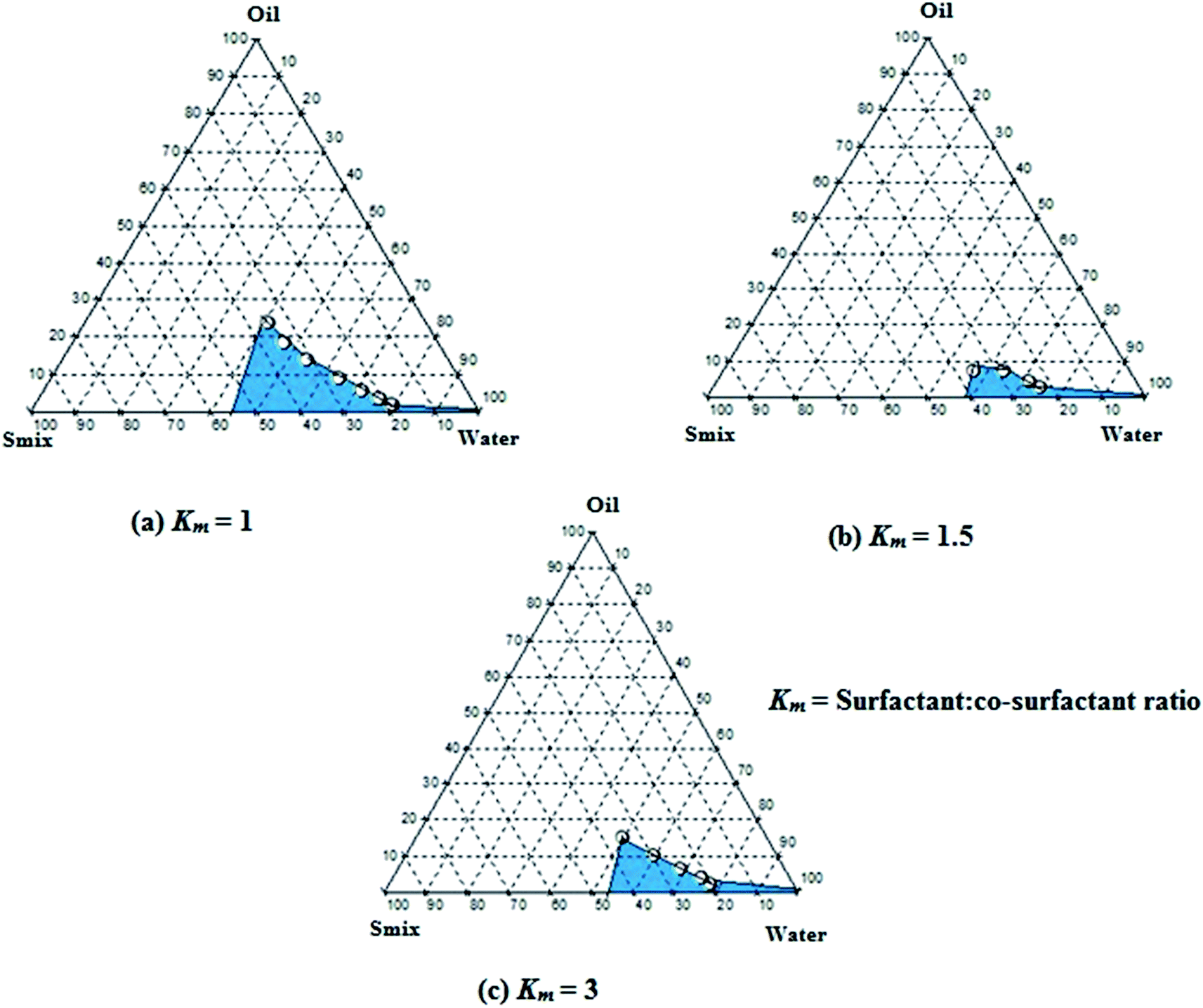 | ||
| Fig. 3 Pseudoternary phase diagrams at (a) Km = 1, (b) Km = 1.5 and (c) Km = 3 indicating o/w microemulsion region. The blue area represents microemulsion existence ranges. | ||
Drug loading efficiency and optimization of formulae
The results of thermodynamic stability studies, dispersibility and drug loading efficiency are presented in (Table 1). It was observed that all five formulations (Curc-E1E_SMEDDS1-5) selected from pseudoternary diagrams passed the thermodynamic stress testing. Therefore, all the formulations were subjected to dispersibility testing. SMEDDS being a thermodynamically stable system readily forms globules after dispersion in a vehicle without phase separation. Upon dilution of SMEDDS formulation, there is every possibility of SMEDDS to separate into different phases, leading to precipitation of a poorly soluble drug.47 When all five formulations were dispersed into aqueous phase, Curc-E1E_SMEDDS 3, 4 and 5 showed spontaneous emulsification and the emulsion so formed was extremely transparent with a bluish tint (Grade A), whereas formulations Curc-E1E_SMEDDS1 and 2 though showed spontaneous emulsification were not so clear (Grade B). Further, all the formulations were subjected to drug loading efficiency studies. Drug loading was in the order of Curc-E1E_SMEDDS5 > Curc-E1E_SMEDDS4 > Curc-E1E_SMEDDS3 > Curc-E1E_SMEDDS2 > Curc-E1E_SMEDDS1 indicating that increase in the quantity of Transcutol HP resulted in increase in the amount of drug entrapped. This may be attributed to the fact that the addition of co-surfactant may lead to greater penetration of the oil phase in the hydrophobic region of the surfactant monomers thereby further decreasing the interfacial tension, which will lead to increase in the fluidity of the interface thus increasing the entropy of the system with increase in drug entapment.39 Highest drug loading (70.52 mg g−1) was found for Curc-E1E_SMEDDS5 containing surfactant:co-surfactant ratio of 1.5:1 and Smix:oil ratio of 8:2. Thus, though the micro-emulsion region of formulation Curc-E1E_SMEDDS5 was lower than the other formulations, loading of drug was found to be very much higher.
| Formulation | Ingredient (mg) | Heating cooling cycle | Centrifugation | Freeze thaw cycle | Dispersibilityb | Drug loading efficiencyc (%) | ||
|---|---|---|---|---|---|---|---|---|
| Heterolipid E1E | Solutol HS15 | Transcutol HP | ||||||
| a ✓ indicates that the formulation passes the test.b Grade A: rapidly forming (within 1 min) emulsion, having a clear or bluish appearance; Grade B: rapidly forming, slightly less clear emulsion, having a bluish white appearance.c Data expressed as mean ± SD (n = 3). | ||||||||
| Curc-E1E_SMEDDS1 | 400 | 600 | — | ✓ | ✓ | ✓ | B | 04.98 ± 1.5 |
| Curc-E1E_SMEDDS2 | 300 | 700 | — | ✓ | ✓ | ✓ | B | 05.0 ± 2.35 |
| Curc-E1E_SMEDDS3 | 300 | 525 | 175 | ✓ | ✓ | ✓ | A | 10.0 ± 2.88 |
| Curc-E1E_SMEDDS4 | 200 | 600 | 200 | ✓ | ✓ | ✓ | A | 15.0 ± 3.34 |
| Curc-E1E_SMEDDS5 | 200 | 480 | 320 | ✓ | ✓ | ✓ | A | 70.52 ± 2.46 |
The solubility of curcumin in developed Curc-E1E_SMEDDS5 was found to be much higher (70.52 ± 2.46 mg g−1) than the previously reported solubility of 21 mg g−1,48 40 mg g−1,30 and 50 mg g−1.28 The total percentage of Smix used to form these SMEDDS were 87.5%, 72.72% and 80% respectively whereas the Smix of formulations reported in this study was around 72.77%, 24.37% and 29.09% respectively to achieve the same curcumin loading. This indicates a remarkable reduction in surfactant and co-surfactant requirements to achieve the same solubility level in the respective SMEDDS. Henceforth, a thermodynamically stable, well dispersible, non-toxic and highly efficient loading batch Curc-E1E_SMEDDS5 was selected. Furthermore, optimization was carried with the help of counter checking the effect of drug loading in Curc-E1E_SMEDDS5 on parameters such as mean globule size, PDI, drug loading and % transmission for 48 h. It can be observed (Table 2) that increasing curcumin loading from 10–50 mg g−1 had no significant influence on mean globule size of Curc-E1E_SMEDDS5 but, there was an increase in mean globule size upon 60 mg g−1 and 70 mg g−1 loading. Percentage transmittance study also suggested that there was decrease in clarity of Curc-E1E_SMEDDS5f and Curc-E1E_SMEDDS5g. This could have happened because of over loading of curcumin resulting into precipitation upon dilution. The drug content was found to be above 99% for formulations Curc-E1E_SMEDDS5a to Curc-E1E_SMEDDS5e. Curc-E1E_SMEDDS5e was selected as an optimized formulation because of small globule size and PDI at highest drug loading amongst all the formulations.
| Formulation | Drug loaded (mg g−1) | Globule size (mean ± SD) | PDI | % transmittance | Drug content (%) |
|---|---|---|---|---|---|
| Curc-E1E_SMEDDS5a | 10 | 21.42 ± 0.49 | 0.21 ± 0.004 | 96.66 ± 0.57 | 99.98 ± 0.13 |
| Curc-E1E_SMEDDS5b | 20 | 22.04 ± 0.68 | 0.25 ± 0.028 | 96.33 ± 1.52 | 99.64 ± 0.50 |
| Curc-E1E_SMEDDS5c | 30 | 22.45 ± 0.50 | 0.23 ± 0.023 | 96.33 ± 1.15 | 99.21 ± 0.40 |
| Curc-E1E_SMEDDS5d | 40 | 21.31 ± 0.29 | 0.23 ± 0.022 | 97 ± 0.00 | 99.82 ± 0.32 |
| Curc-E1E_SMEDDS5e | 50 | 22.73 ± 0.45 | 0.24 ± 0.032 | 97 ± 0.00 | 99.22 ± 0.27 |
| Curc-E1E_SMEDDS5f | 60 | 128 ± 2.04 | 0.74 ± 0.077 | 79 ± 1.0 | 84.40 ± 1.64 |
| Curc-E1E_SMEDDS5g | 70 | 156 ± 1.52 | 0.82 ± 0.049 | 72.66 ± 1.15 | 73.48 ± 1.89 |
Characterization of Curc-E1E_SMEDDS5e
 | ||
| Fig. 4 TEM micrograph of Curc-E1E_SMEDDS5e (scale bar: 200 nm). | ||
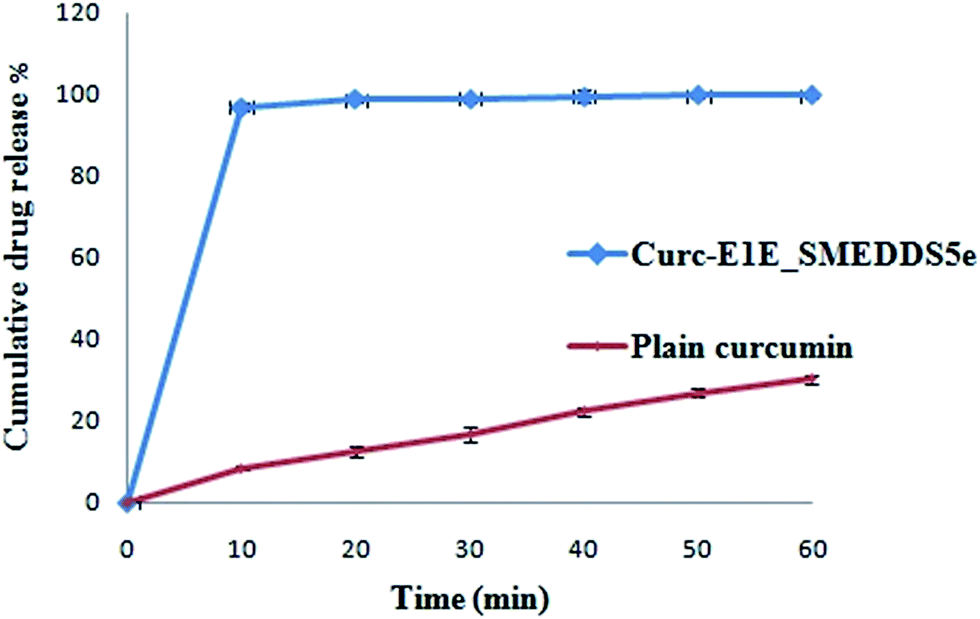 | ||
| Fig. 5 Release profiles of curcumin as pure drug and Curc-E1E_SMEDDS5e in USP buffer of pH 1.2 using USP paddle (100 rpm) apparatus at 37 °C, data expressed as mean ± SD (n = 3). | ||
| Formulation | Cmax (μg mL−1) | Tmax (min) | AUC0–∞ (μg h mL−1) |
|---|---|---|---|
| Curc-E1E_SMEDDS5e | 4.921 ± 0.42 | 60 | 17.30 ± 1.50 |
| Curcumin aqueous suspension | 0.186 ± 0.006 | 60 | 0.234 ± 0.023 |
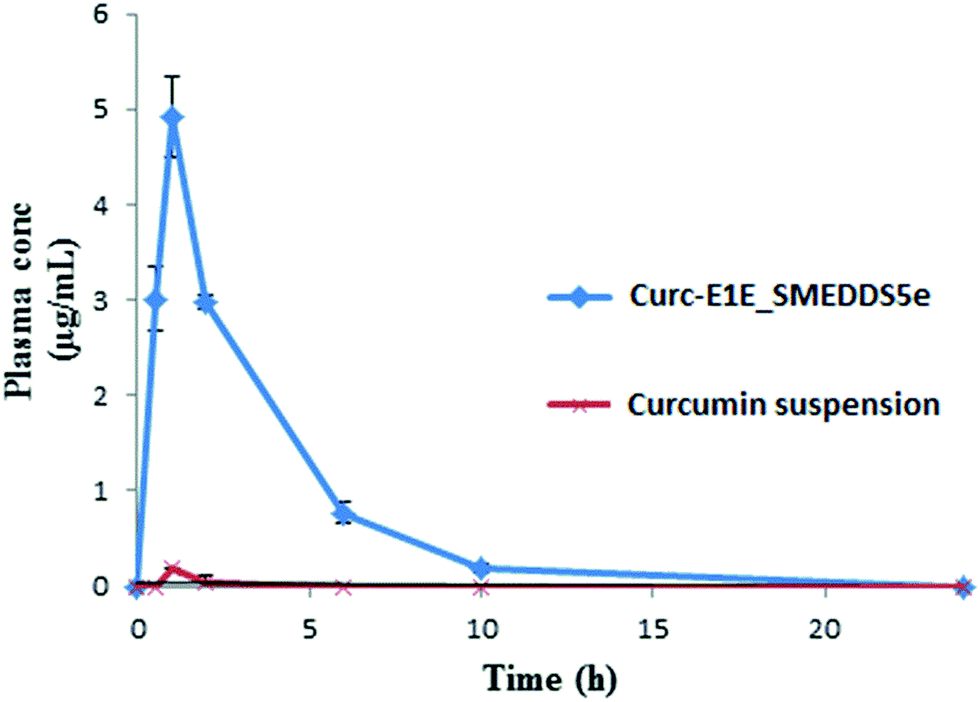 | ||
| Fig. 6 The mean concentration–time curve after a single oral dose (50 mg kg−1) of curcumin aqueous suspension and Curc-E1E_SMEDDS5e (n = 3, mean ± SD). | ||
In vitro anticancer activity. SRB assay revealed that Curc-E1E_SMEDDS5e had strong anticancer activity as indicated by GI50 and TGI values comparable to doxorubicin (GI50 and TGI < 10 μg mL−1) (Fig. 7). The LC50 value for Curc-E1E_SMEDDS5e was found to be 71.6 μg mL−1 revealing that the toxicity level was practically none. The results of in vitro anticancer activity suggest that Curc-E1E_SMEDDS5e is a potent anticancer formulation with practically no cytotoxicity as compared to doxorubicin.
 | ||
| Fig. 7 Growth curve of human cervix cancer cell line HeLa. Data reported are means ± SD (n = 3). | ||
Antimicrobial activity. The MICs of Curc-E1E_SMEDDS5e against E. coli and S. aureus were 48.62 μg mL−1 and 97.65 μg mL−1 respectively whereas plain curcumin dispersed in water showed MIC values >500 μg mL−1 against both the bacterial strains. A higher MIC value of dispersed curcumin was obvious, since curcumin has poor solubility in water limiting its bioavailability. Moreover, antimicrobial activity of control formulation without curcumin was performed and it was observed that there was no growth inhibition of E. coli and S. aureus. This confirmed that all the activities showed were due to curcumin alone. Therefore, from this study it can be concluded that there was a great enhancement of antimicrobial efficacy of curcumin upon its solubilization in micro droplet form using a novel heterolipid E1E.
Stability studies. Curc-E1E_SMEDDS5e formulations were evaluated under three different conditions for three months (Table 4) as per ICH guidelines and it was observed that there was no significant change in the mean globule size, PDI and drug content. Upon visual observation Curc-E1E_SMEDDS5e appeared as transparent and homogenous liquid. Throughout the entire period of the study, Curc-E1E_SMEDDS5e was evaluated for dispersibility test and it was observed that it dispersed within the 10 seconds forming clear, transparent, micro-emulsion at all conditions. The formulation showed no signs of destabilization, could withstand accelerated condition and with no signs of precipitation.
| Storage condition time (days) | Particle size (nm) | Polydispersity index (PDI) | % drug content | Dispersibilitya |
|---|---|---|---|---|
| a Grade A: rapidly forming (within 1 min) emulsion, having a clear or bluish appearance. | ||||
| 25 ± 2 °C/60 ± 5% RH | ||||
| 30 | 22.39 ± 0.20 | 0.243 ± 0.010 | 100.25 ± 1.49 | A |
| 60 | 22.97 ± 4.60 | 0.227 ± 0.052 | 100.20 ± 1.40 | A |
| 90 | 21.98 ± 4.82 | 0.245 ± 0.060 | 99.62 ± 0.90 | A |
|
||||
| 30 ± 2 °C/65 ± 5% RH | ||||
| 30 | 23.14 ± 2.31 | 0.212 ± 0.274 | 99.2 ± 1.09 | A |
| 60 | 22.75 ± 4.32 | 0.220 ± 0.030 | 99.12 ± 1.60 | A |
| 90 | 21.52 ± 2.69 | 0.254 ± 0.023 | 98.96 ± 1.10 | A |
|
||||
| 45 ± 2 °C/75 ± 5% RH | ||||
| 30 | 22.88 ± 2.19 | 0.215 ± 0.26 | 100.53 ± 1.90 | A |
| 60 | 23.56 ± 3.11 | 0.254 ± 0.32 | 99.48 ± 1.45 | A |
| 90 | 24.87 ± 2.75 | 0.280 ± 0.42 | 98.13 ± 1.21 | A |
In summary, solubility data showed that the novel heterolipid E1E enhanced the solubility of curcumin than parent oleic acid and ethyl oleate, and was successfully utilized for the formulation development of stable curcumin SMEDDS. The developed Curc-E1E_SMEDDS5e displayed very clear, transparent and stable microemulsion with an average globule size of 22.39 nm and PDI of 0.243. The in vitro dissolution studies showed that 97% of curcumin was released from Curc-E1E_SMEDDS5e within 10 min as compared to only 8% of plain curcumin. Developed SMEDDS was found to have very high entrapment efficiency of curcumin (70.52 mg g−1) as compare to reported 50 mg g−1, 40 mg g−1 and 21 mg g−1 by different researchers. Pharmacokinetic study in rats revealed that optimized formulation, Curc-E1E_SMEDDS5e, showed 26.45-fold greater absorption, as compared to curcumin administered as aqueous suspension at same oral dose of 50 mg kg−1. The pharmacokinetic parameter, Cmax was very high (4.921 μg mL−1) compared to Cmax of curcumin aqueous suspension (0.186 μg mL−1) reflective of greater bioavailability of curcumin from Curc-E1E_SMEDDS5e. Dilution and accelerated tests found no effect on globule size. The potent anticancer and antimicrobial activity exhibited by Curc-E1E_SMEDDS5e was in agreement with the observed enhancement in the bioavailability and permeation. In addition, Curc-E1E_SMEDDS5e was stable over the studied time periods and storage conditions. In conclusion the developed Curc-E1E_SMEDDS5e using a novel heterolipid E1E as an oil phase can be used as a potential effective dosage form in nutraceuticals and food products due to enhanced solubility and bioavailability of curcumin.
Conflict of interest
The authors report no conflicts of interest.Acknowledgements
Dinesh M. Dhumal and Priya Kothari are thankful to the Department of Science and Technology and University Grants Commission, Government of India for financial support. The authors acknowledge Advanced Center for Treatment Research and Education in Cancer (ACTREC), Mumbai, India for SRB assay.References
- R. K. Basniwal, H. S. Buttar, V. K. Jain and N. Jain, Curcumin Nanoparticles: Preparation, Characterization, and Antimicrobial Study, J. Agric. Food Chem., 2011, 59, 2056–2061 CrossRef PubMed.
- B. T. Kurien, A. D'Souza and R. H. Scofield, Heat-solubilized curry spice curcumin inhibits antibody–antigen interaction in in vitro studies: a possible therapy to alleviate autoimmune disorders, Mol. Nutr. Food Res., 2010, 54, 1202–1209 CAS.
- R. K. Maheshwari, A. K. Singh, J. Gaddipati and R. C. Srimal, Multiple biological activities of curcumin: a short review, Life Sci., 2006, 78, 2081–2087 CrossRef CAS PubMed.
- P. Basnet and N. Skalko-Basnet, Curcumin: an anti-inflammatory molecule from a curry spice on the path to cancer treatment, Molecules, 2011, 16, 4567–4598 CrossRef CAS PubMed.
- J. Wang, H. Wang, R. Zhu, Q. Liu, J. Fei and S. Wang, Anti-inflammatory activity of curcumin-loaded solid lipid nanoparticles in IL-1β transgenic mice subjected to the lipopolysaccharide-induced sepsis, Biomaterials, 2015, 53, 475–483 CrossRef CAS PubMed.
- P. B. Memvanga, R. Coco and V. Préat, An oral malaria therapy: Curcumin-loaded lipid-based drug delivery systems combined with β-arteether, J. Controlled Release, 2013, 172, 904–913 CrossRef CAS PubMed.
- B. B. Aggarwal and K. B. Harikumar, Potential therapeutic effects of curcumin, the anti-inflammatory agent, against neurodegenerative, cardiovascular, pulmonary, metabolic, autoimmune and neoplastic diseases, Int. J. Biochem. Cell Biol., 2009, 41, 40–59 CrossRef CAS PubMed.
- P. Anand, A. B. Kunnumakkara, R. A. Newman and B. B. Aggarwal, Bioavailability of Curcumin: Problems and Promises, Mol. Pharm., 2007, 4, 807–818 CrossRef CAS PubMed.
- B. B. Aggarwal and B. Sung, Pharmacological basis for the role of curcumin in chronic diseases: an age-old spice with modern targets, Trends Pharmacol. Sci., 2009, 30, 85–94 CrossRef CAS PubMed.
- L. Mayol, C. Serri, C. Menale, S. Crispi, M. T. Piccolo, L. Mita, S. Giarra, M. Forte, A. Saija, M. Biondi and D. G. Mita, Curcumin loaded PLGA–poloxamer blend nanoparticles induce cell cycle arrest in mesothelioma cells, Eur. J. Pharm. Biopharm., 2015, 93, 37–45 CrossRef CAS PubMed.
- G. Shoba, D. Joy, T. Joseph, M. Majeed, R. Rajendran and P. S. S. R. Srinivas, Influence of Piperine on the Pharmacokinetics of Curcumin in Animals and Human Volunteers, Planta Med., 1998, 64, 353–356 CrossRef CAS PubMed.
- H. Mu, R. Holm and A. Müllertz, Lipid-based formulations for oral administration of poorly water-soluble drugs, Int. J. Pharm., 2013, 453, 215–224 CrossRef CAS PubMed.
- Y. Kawabata, K. Wada, M. Nakatani, S. Yamada and S. Onoue, Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: basic approaches and practical applications, Int. J. Pharm., 2011, 420, 1–10 CrossRef CAS PubMed.
- S. Bisht, G. Feldmann, S. Soni, R. Ravi, C. Karikar, A. Maitra and A. Maitra, Polymeric nanoparticle-encapsulated curcumin (“nanocurcumin”): a novel strategy for human cancer therapy, J. Nanobiotechnol., 2007, 5, 1–18 CrossRef PubMed.
- M. Takahashi, S. Uechi, K. Takara, Y. Asikin and K. Wada, Evaluation of an oral carrier system in rats: bioavailability and antioxidant properties of liposome-encapsulated curcumin, J. Agric. Food Chem., 2009, 57, 9141–9146 CrossRef CAS PubMed.
- C. Gonçalves, P. Pereira, P. Schellenberg, P. J. Coutinho and F. M. Gama, Self-Assembled Dextrin Nanogel as Curcumin Delivery System, J. Biomater. Nanobiotechnol., 2012, 3, 178–184 CrossRef.
- V. Kakkar, S. K. Muppu, K. Chopra and I. P. Kaur, Curcumin loaded solid lipid nanoparticles: an efficient formulation approach for cerebral ischemic reperfusion injury in rats, Eur. J. Pharm. Biopharm., 2013, 85, 339–345 CrossRef CAS PubMed.
- R. Yang, S. Zhang, D. Kong, X. Gao, Y. Zhao and Z. Wang, Biodegradable polymer–curcumin conjugate micelles enhance the loading and delivery of low-potency curcumin, Pharm. Res., 2012, 29, 3512–3525 CrossRef CAS PubMed.
- K. Maiti, K. Mukherjee, A. Gantait, B. P. Saha and P. K. Mukherjee, Curcumin–phospholipid complex: preparation, therapeutic evaluation and pharmacokinetic study in rats, Int. J. Pharm., 2007, 330, 155–163 CrossRef CAS PubMed.
- S. Mourtas, A. N. Lazar, E. Markoutsa, C. Duyckaerts and S. G. Antimisiaris, Multifunctional nanoliposomes with curcumin–lipid derivative and brain targeting functionality with potential applications for Alzheimer disease, Eur. J. Med. Chem., 2014, 80, 175–183 CrossRef CAS PubMed.
- K. Coradini, F. O. Lima, C. M. Oliveira, P. S. Chaves, M. L. Athayde, L. M. Carvalho and R. C. R. Beck, Co-encapsulation of resveratrol and curcumin in lipid-core nanocapsules improves their in vitro antioxidant effects, Eur. J. Pharm. Biopharm., 2014, 88, 178–185 CrossRef CAS PubMed.
- A. L. Palange, D. Di Mascolo, C. Carallo, A. Gnasso and P. Decuzzi, Lipid–polymer nanoparticles encapsulating curcumin for modulating the vascular deposition of breast cancer cells, Nanomedicine: Nanotechnology, Biology and Medicine, 2014, 10, 991–1002 CrossRef CAS PubMed.
- K. Vandita, B. Shashi, K. G. Santosh and K. I. Pal, Enhanced Apoptotic Effect of Curcumin Loaded Solid Lipid Nanoparticles, Mol. Pharmaceutics, 2012, 9, 3411–3421 CrossRef CAS PubMed.
- N. Sermkaew, W. Ketjinda, P. Boonme, N. Phadoongsombut and R. Wiwattanapatapee, Liquid and solid self-microemulsifying drug delivery systems for improving the oral bioavailability of andrographolide from a crude extract of Andrographis paniculata, Eur. J. Pharm. Sci., 2013, 50, 459–466 CrossRef CAS PubMed.
- D. W. Kim, M. S. Kwon, A. M. Yousaf, P. Balakrishnan, J. H. Park, D. S. Kim, B.-J. Lee, Y. J. Park, C. S. Yong, J. O. Kim and H.-G. Choi, Comparison of a solid SMEDDS and solid dispersion for enhanced stability and bioavailability of clopidogrel napadisilate, Carbohydr. Polym., 2014, 114, 365–374 CrossRef CAS PubMed.
- Q. Li, W. Zhai, Q. Jiang, R. Huang, L. Liu, J. Dai, W. Gong, S. Du and Q. Wu, Curcumin–piperine mixtures in self-microemulsifying drug delivery system for ulcerative colitis therapy, Int. J. Pharm., 2015, 490, 22–31 CrossRef CAS PubMed.
- A. Grill, B. Koniar and J. Panyam, Co-delivery of natural metabolic inhibitors in a self-microemulsifying drug delivery system for improved oral bioavailability of curcumin, Drug Delivery Transl. Res., 2014, 4, 344–352 CrossRef CAS PubMed.
- X. Wu, J. Xu, X. Huang and C. Wen, Self-microemulsifying drug delivery system improves curcumin dissolution and bioavailability, Drug Dev. Ind. Pharm., 2011, 37, 15–23 CrossRef CAS PubMed.
- L. Hu, Y. Jia, F. Niu, Z. Jia, X. Yang and K. Jiao, Preparation and Enhancement of Oral Bioavailability of Curcumin Using Microemulsions Vehicle, J. Agric. Food Chem., 2012, 60, 7137–7141 CrossRef CAS PubMed.
- S. Setthacheewakul, S. Mahattanadul, N. Phadoongsombut, W. Pichayakorn and R. Wiwattanapatapee, Development and evaluation of self-microemulsifying liquid and pellet formulations of curcumin, and absorption studies in rats, Eur. J. Pharm. Biopharm., 2010, 76, 475–485 CrossRef CAS PubMed.
- L. Zhang, W. Zhu, C. Yang, H. Guo, A. Yu, J. Ji, Y. Gao, M. Sun and G. Zhai, A novel folate-modified self-microemulsifying drug delivery system of curcumin for colon targeting, Int. J. Nanomed., 2012, 7, 151–162 CAS.
- C. W. Pouton and C. J. Porter, Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies, Adv. Drug Delivery Rev., 2008, 60, 625–637 CrossRef CAS PubMed.
- P. P. Constantinides and J.-P. Scalart, Formulation and physical characterization of water-in-oil microemulsions containing long-versus medium-chain glycerides, Int. J. Pharm., 1997, 158, 57–68 CrossRef CAS.
- S. D. Mandawgade, S. Sharma, S. Pathak and V. B. Patravale, Development of SMEDDS using natural lipophile: application to β-artemether delivery, Int. J. Pharm., 2008, 362, 179–183 CrossRef CAS PubMed.
- R. S. Kalhapure and K. G. Akamanchi, Oleic acid based heterolipid synthesis, characterization and application in self-microemulsifying drug delivery system, Int. J. Pharm., 2012, 425, 9–18 CrossRef CAS PubMed.
- R. S. Kalhapure and K. G. Akamanchi, Oleodendrimers: A novel class of multicephalous heterolipids as chemical penetration enhancers for transdermal drug delivery, Int. J. Pharm., 2013, 454, 158–166 CrossRef CAS PubMed.
- D. Gugulothu, P. Desai and V. Patravale, A Versatile Liquid Chromatographic Technique for Pharmacokinetic Estimation of Curcumin in Human Plasma, J. Chromatogr. Sci., 2014, 52, 872–879 Search PubMed.
- V. Bali, M. Ali and J. Ali, Nanocarrier for the enhanced bioavailability of a cardiovascular agent: in vitro, pharmacodynamic, pharmacokinetic and stability assessment, Int. J. Pharm., 2011, 403, 46–56 CrossRef CAS PubMed.
- S. Shafiq, F. Shakeel, S. Talegaonkar, F. J. Ahmad, R. K. Khar and M. Ali, Development and bioavailability assessment of ramipril nanoemulsion formulation, Eur. J. Pharm. Biopharm., 2007, 66, 227–243 CrossRef CAS PubMed.
- R. Parveen, S. Baboota, J. Ali, A. Ahuja, S. S. Vasudev and S. Ahmad, Oil based nanocarrier for improved oral delivery of silymarin: in vitro and in vivo studies, Int. J. Pharm., 2011, 413, 245–253 CrossRef CAS PubMed.
- V. Vichai and K. Kirtikara, Sulforhodamine B colorimetric assay for cytotoxicity screening, Nat. Protoc., 2006, 1, 1112–1116 CrossRef CAS PubMed.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, New colorimetric cytotoxicity assay for anticancer-drug screening, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS PubMed.
- ICH guidelines Q 1 A (R2), Stability testing of new drug substances and products, 2003.
- P. Hyma, J. Noor, Raheemunissa, G. Sreelekha and K. Babu, Microemulsion based hydrogel formulation for topical delivery, Pharmanest, 2014, 5, 1874–1880 Search PubMed.
- U. Gupta, H. B. Agashe, A. Asthana and N. Jain, Dendrimers: novel polymeric nanoarchitectures for solubility enhancement, Biomacromolecules, 2006, 7, 649–658 CrossRef CAS PubMed.
- V. Borhade, H. Nair and D. Hegde, Design and Evaluation of Self-Microemulsifying Drug Delivery System (SMEDDS) of Tacrolimus, AAPS PharmSciTech, 2008, 9, 13–21 CrossRef CAS PubMed.
- M. J. Qureshia, C. Mallikarjun and W. G. Kian, Enhancement of solubility and therapeutic potential of poorly soluble lovastatin by SMEDDS formulation adsorbed on directly compressed spray dried magnesium aluminometasilicate liquid loadable tablets: a study in diet induced hyperlipidemic rabbits, Asian J. Pharm. Sci., 2015, 10, 40–56 CrossRef PubMed.
- J. Cui, B. Yu, Y. Zhao, W. Zhu, H. Li, H. Lou and G. Zhai, Enhancement of oral absorption of curcumin by self-microemulsifying drug delivery systems, Int. J. Pharm., 2009, 371, 148–155 CrossRef CAS PubMed.
- Y. P. Wang, Y. Gan and X. X. Zhang, Novel gastroretentive sustained-release tablet of tacrolimus based on self-microemulsifying mixture: in vitro evaluation and in vivo bioavailability test, Acta Pharmacol. Sin., 2011, 32, 1294–1302 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra18112g |
| This journal is © The Royal Society of Chemistry 2015 |