
The use of polymer-supported Candida antarctica lipase B to achieve the entropically-driven ring-opening polymerization of macrocyclic bile acid derivatives via transesterification: selectivity of the reactions and the structures of the polymers produced†
Philip Hodge* and
Abdel B. Chakiri
Department of Chemistry, University of Manchester, Oxford Road, Manchester M13 9PL, UK. E-mail: Philip.Hodge@manchester.ac.uk; a.b.chakiri@gmail.com; Tel: +44 (0)1524 791728
First published on 20th October 2015
Abstract
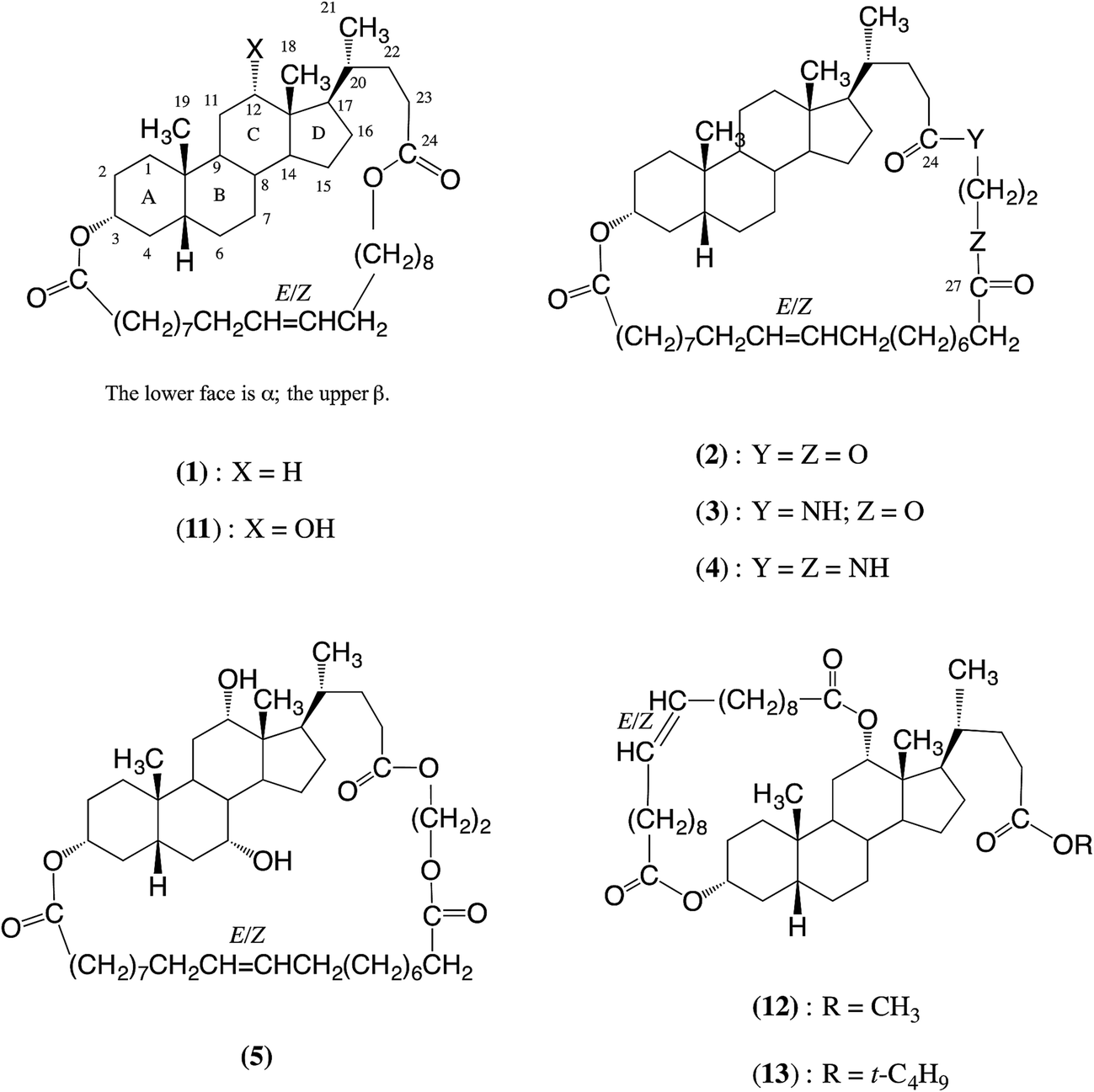
Various macrocyclic lactones containing bile acid residues successfully undergo entropically-driven ring-opening polymerization via transesterifications (TEs) catalyzed by polymer-supported Candida antarctica lipase B (PS-CALB) when the 3α- and 24-positions are linked by C14 or C20 chains or by a 23-atom chain, but not when the 3α- and 12α-positions are linked by a C20 chain. This suggests that the α-face of the bile acid derivatives in the region of the A–C rings sit at the active site of the enzyme. TEs of model bile acid derivatives indicate that the order of reactivity of ester groups catalyzed by CALB is 3α-OAc > 26-OAc ≫ 24-CO2R, though the latter do react. The 12α-OH does not react. Accordingly, under the polymerization conditions used in this work, the polymeric products formed from the macrocyclic bile acid derivatives containing a C14 or C20 linkage between the 3α- and 24-positions are expected to be a polymer chain formed essentially by ring-opening at the 3α-position, with the repeat units arranged in a regular head-to-tail manner. This differs from the structure of the polymers obtained by ED-ROMP of the same macrocycles. The macrocycles formed formally from glycol lithocholate (the diol unit) and E/Z-eicos-10-enedioic acid (the diacid unit) afford polymers that essentially have these units linked in an alternating manner. There are regiosiomers because of the unsymmetrical diol unit. Esterifications and TEs catalyzed by PS-CALB using a large excess of appropriate volatile aliphatic esters are a useful alternative to more expensive activated esters in the synthesis of esters using PS-CALB.
Introduction
Entropically-driven ring-opening polymerization (ED-ROP) is a relatively new method of polymer synthesis.1,2 The reactions are usually based on ring:chain equilibria (RCE), i.e. the well-known equilibria that can exist, under appropriate reaction conditions, between a polymer and the corresponding family of macrocyclic oligomers (MCOs): see Scheme 1.1,3 The position of the RCE is very dependent on the concentration of the reactants and if MCOs are taken at high concentration and the RCE established, polymer is formed in high yield. Such polymerizations have several attractive features.1,2 For example, they are essentially thermally neutral, they proceed without the formation of any byproducts, they extend the range of application of ROPs to large cyclics which may, therefore, contain substantial structural features in the rings such as steroid moieties, and if few end groups are present in the reaction system the molecular weights obtained can be unusually high.4–6 Essentially ED-ROP is a green method of polymer synthesis.1,7 If used in combination with cyclodepolymerization (CDP) to prepare the MCOs it offers the prospect of becoming a useful means of recycling various condensation polymers.1,8 | ||
| Scheme 1 Scheme for a generalized Ring:Chain Equilibrium (RCE). | ||
Biocompatible and biodegradable polymeric materials have many potential applications, for example, as sutures, stents, tissue engineering scaffolds, drug delivery systems or bone screws.9 The polymers are of particular interest if, when they biodegrade, they give compounds that the organism can excrete easily or is capable of transforming into harmless metabolites. Recently, in this context, Zhu et al. have studied polymers prepared from bile acid derivatives,5,6,10,11 often in combination with fatty acid derivatives.5,6 Simple hydrocarbon chains are very flexible, but bile acid moieties introduce a degree of stiffness to the chains so giving potentially more useful materials, e.g. materials with Tgs greater than body temperature.
Zhu et al. have prepared pure macrocycles 1 (35 ring atoms),5,6 and 2–5 (38 ring atoms),6 see Chart 1, then carried out entropically-driven ring-opening metathesis polymerizations (ED-ROMPs) using Grubbs second generation catalyst (G2). These polymerizations gave polymers 6–10 respectively: see Chart 2. Very recently the work has been extended to the ROMPs of MCOs of 11 (35 ring atoms per repeat unit), 12 and 13 (each 29 ring atoms per repeat unit).12 The products are polymers 14–16. Copolymers have also been prepared.12,13 It should be noted that all these polymers have unsymmetrical repeat units and so are expected to have the units present in head-to-head, head-to-tail and tail-to-tail arrangements. A problem with ROMPs is that the products are sometimes discoloured by traces of ruthenium residues.
 | ||
| Chart 1 Macrocyclic bile acid derivatives that have been successfully subjected to ROMP. | ||
 | ||
| Chart 2 Structures of polymers formed by ROMP of the macrocycles shown in Chart 1. | ||
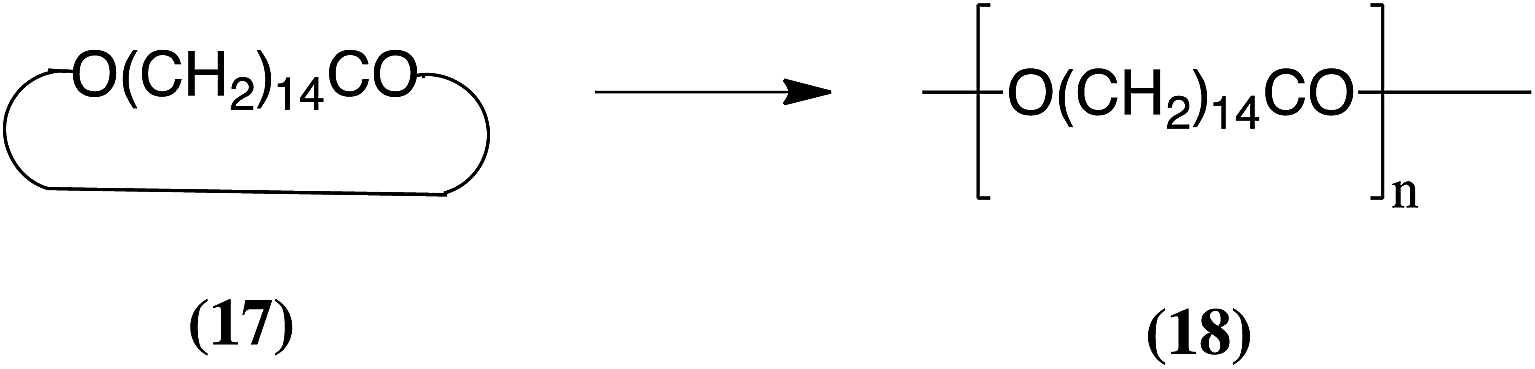
If macrocycles contain ester linkages in the rings an alternative method of carrying out ED-ROPs is via transesterifications (TEs). Several such ED-ROPs of medium-sized lactones catalyzed by polymer-supported Candida antarctica lipase B (PS-CALB) were reported several years ago.14,15 An important example is the ED-ROP of pentadecanolactone (17) because the polymeric product 18 has many material properties similar to those of polyethylene.15
PS-CALB is commercially available and when CALB is used in this form the catalyst is easily filtered off at end of the polymerization, and can possibly be reused.16 In many biological applications, any enzyme that might remain in the product will almost certainly be biodegraded. Accordingly, ED-ROPs using CALB are an attractive type of polymerization in the context of biocompatible polymers.
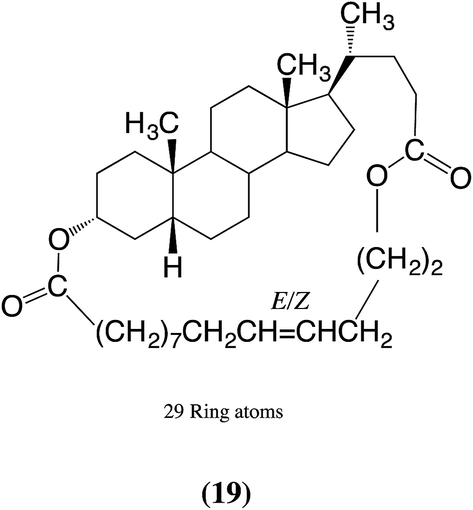
Since the early work with PS-CALB much larger lactones, some with as many as 84 ring atoms, have been shown to undergo ED-ROP successfully.17 These include macrocycles 1 and 19, which gave polymeric products with Mn values of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 and 18200, respectively, and Mw of 49500 and 32600.17 Whilst these values are significantly less than some of those reported for the ROMP reactions mentioned above,5,6 they are nevertheless suitable for many applications. Very recently Zhu et al. reported a further example of an ED-ROP of a macrocyclic bile acid derivative catalyzed by PS-CALB, namely that of macrocycle 5.18 In a typical experiment the polymeric product (49% yield after reprecipitation) had Mn 20200 and Mw 26300.18
400 and 18200, respectively, and Mw of 49500 and 32600.17 Whilst these values are significantly less than some of those reported for the ROMP reactions mentioned above,5,6 they are nevertheless suitable for many applications. Very recently Zhu et al. reported a further example of an ED-ROP of a macrocyclic bile acid derivative catalyzed by PS-CALB, namely that of macrocycle 5.18 In a typical experiment the polymeric product (49% yield after reprecipitation) had Mn 20200 and Mw 26300.18
In this paper we first report the results an investigation of further ED-ROPs of macrocyclic bile acid derivatives achieved via TEs catalyzed by PS-CALB. These were carried out to explore further the scope of these green polymerizations. We then report TE studies of various model bile acid derivatives. These studies were carried out to determine the selectivity of PS-CALB in its reaction with these derivatives. The results allow us to predict the structures of the various polymeric products. Where appropriate we use our results to rationalize earlier published results.17,18
Before discussing the present results it is important to consider what is already known about CALB in the present context. Its main role in vivo is to catalyze the synthesis and degradation of fatty acid triglycerides. It achieves this using a classical serine–histidine–aspartic acid triad.14,19,20 CALB first catalyzes the transfer of an acyl group from an ester in solution onto the hydroxyl group of the serine moiety: see Scheme 2, Reaction (1), for the case of a lactone. This activates the acyl group for further transfer to an hydroxyl group. In the initiation process the acylated serine reacts with water to give an ω-hydroxyacid: see Reaction (2). The ω-hydroxyacid now reacts as an alcohol with a further activated acyl group to form a dimer (Reaction (3)). Reaction (1) followed by Reaction (3) now repeats (Propagation) to build up first an oligomer, then a polymer. It is clear from this that CALB can catalyze both acylations of alcohols and transesterifications of esters, and that the selectivity for both reactions is expected to be the same.
 | ||
Scheme 2 Scheme of catalysis by CALB, using a lactone as the monomer.  represents the serine alcohol group of CALB. The alcohol produced by the Initiation (Reaction (2)) takes part in Reaction (3), so on each cycle the Propagation Reaction (3) adds one more hydroxy-acid unit to the hydroxy end of the polymer chain. represents the serine alcohol group of CALB. The alcohol produced by the Initiation (Reaction (2)) takes part in Reaction (3), so on each cycle the Propagation Reaction (3) adds one more hydroxy-acid unit to the hydroxy end of the polymer chain. | ||
In previously published work with bile acid derivatives it has been shown that 3α-hydroxyl groups are acetylated efficiently when treated with CALB and vinyl acetate in tetrahydrofuran.21,22 No reaction occurs with 7α-, 7β- and 12α-hydroxyl groups: see formula 1 for the numbering of bile acid derivatives.21–23 The 3α-hydroxyl group of methyl cholate (20) is also selectively acylated when treated with various methyl or vinyl alkanoates and CALB.22 Interestingly, CO2CH3 groups at the 24-position, which are present in many of the substrates studied, are not reported to react.21,22 They do, however, react under more vigorous conditions since when cholic acid (21) is treated with a relatively large amount of CALB in tetrahydrofuran at 50 °C for 7 days oligomerization occurs, i.e. the 3α-hydroxyl groups and 24CO2H groups of different molecules react together.23 With other steroid families, CALB catalyses the acylation, using various methyl alkanoates, of the 21-hydroxyl group in hydrocortisone (22),21,24 and similar steroids,21 but not the 11β- or 17α-hydroxyl groups.21,24 Androstane 23 reacts with octan-1-ol to achieve selectively the deacetylation of the 16β-acetate.25 It is clear from these results that, with the types of compounds investigated, CALB only successfully catalyses esterifications or transesterifications at the 3α-, 16β- and 21-positions, i.e. groups near the ends of the rigid tetracyclic steroid nucleus. Alcohol or ester groups at the 7α-, 7β-, 11β-, 12α-, and 17α-positions do not react.


Results and discussion
It is clear from the above discussion that the present work falls into three main parts. First, an investigation of the reactions of further macrocycles which can potentially undergo ED-ROP with PS-CALB. Then, an investigation of various TEs of model bile acid derivatives carried out to probe the selectivity of CALB. Finally, use of the above results to rationalize why certain macrocycles polymerize whilst others do not, and to allow the structures of the polymeric products to be predicted. At appropriate places we also use our results to rationalize earlier published results.17,18Further ED-ROP reactions
Four macrocycles were available to us that have not been reacted with PS-CALB before. These are macrocycles 2, 11, 12 and 13.6,12 ED-ROPs of these using PS-CALB as catalyst were now attempted. Though macrocycle 1 has been studied before,17 it was included here to facilitate comparisons. The results are summarized in Table 1.| Entry | MCO(s) | Reaction time (h) | Yield (%) | Molecular weightsb | Đc | |
|---|---|---|---|---|---|---|
| Mn | Mw | |||||
| a See Experimental for standard procedure. Reaction times are as shown.b By SEC relative to polystyrene standards.17c Đ = dispersity. See ref. 26 for IUPAC's recent recommendations on dispersity.d The PS-CALB from the experiment summarized in the preceding entry was collected, washed with dry toluene and then reused.e No polymer peak was detected. SEC indicated the macrocycles were recovered. | ||||||
| 1 | 1 | 20 | 95 | 19300 |
31700 |
1.83 |
| 2 | 1 | 20d | 90 | 18200 |
32200 |
1.77 |
| 3 | 1 | 20d | 93 | 18600 |
34100 |
1.83 |
| 4 | 1 | 10 | 89 | 18500 |
32000 |
1.73 |
| 5 | 2 | 20 | 94 | 20500 |
46500 |
2.23 |
| 6 | 11 | 20 | 92 | 15300 |
30700 |
2.01 |
| 7 | 11 | 10 | 90 | 11300 |
20700 |
1.83 |
| 8 | 12 | 20 | 98 | —e | —e | — |
| 9 | 13 | 20 | 96 | —e | —e | — |
The polymerizations were first attempted under standard conditions that were essentially the same as those used by us previously.17 Thus, an 18% w/v solution of the substrate in toluene at 70 °C was treated with PS-CALB (18% the weight of the substrate) for 20 h. At the end of the reaction period the polymer beads were filtered off, the solvent removed from the filtrate under vacuum and the concentrate added dropwise into ice cold methanol. Any polymeric product precipitated, and was collected, dried and characterized by 1H NMR and FT-IR spectroscopies and by analytical size exclusion chromatography (SEC). The results are summarized in Table 1. Typical data is included in the ESI.†
The polymerizations of macrocycles 1, 2 and 11 took place smoothly to give 90–95% yields of the corresponding polymers: see entries 1–3, 5 and 6. The polymers were obtained as clear gums, soluble in chloroform, with satisfactory FT-IR and 1H NMR spectra. The 1H NMR spectrum of the product from 11 had the characteristic one-proton signal at δ 3.95 ppm due to the β-proton on the carbon bearing the 12α-hydroxyl group. Had this hydroxyl been acylated the signal would have shifted to near δ 5.1 ppm. No such signal was detected, so, as expected, acylation of the 12α-hydroxyl did not occur.
The polymeric products from macrocycle 1, Table 1 entries 1–3, gave products with molecular weights very similar to those obtained previously.17 The results indicate that the PS-CALB can be reused at least twice. Macrocycles 2 and 11 gave polymers with comparable molecular weights: see Table 1, entries 5 and 6. Since by SEC MCOs 11 contained only 85% of the cyclic monomer, a 92% yield of polymer indicates that the cyclic dimer (70-ring atoms; 13%) must also have reacted.12
The results of polymerizing macrocycles 1 and 11 using a reaction time of only 10 h indicate, compare Table 1 entries 1 and 4, and 6 and 7, that it is not necessary to use a reaction time of 20 h, as used in the standard conditions, to obtain polymers with molecular weights >10000 (see later in Section 3).
The most interesting result is that MCOs 12 and 13 did not polymerize at all, see entries 8 and 9: the macrocycles were simply recovered. This result was confirmed by repeating the experiments. It will be recalled that both of these MCOs underwent ED-ROMP successfully when treated with G2.12
To conclude this part, the above results confirm that the 12α-hydroxyl group does not react but that it does not prevent the steroid derivative 11 from binding to the CALB active site to achieve a successful ED-ROP: see Table 1, entries 6 and 7. However, increasing the bulk on the α-face by the acylation of the 12α-hydroxyl group prevents satisfactory binding to the active site of CALB: see entries 8 and 9. Whereas the 20-carbon linkage between the 3α- and 12α-positions, as in 12 and 13, prevents polymerization, a 20-carbon linkage or a 23-atom chain between the 3α- and 24-positions, as in 1, 2, 5 and 11, does not. Presumably the latter linkages do not screen the α-face of the A–C rings of the steroid: see formula 1 for the labeling of the rings. As reported previously,17 using a shorter 14-carbon chain between the 3α- and 24-positions, as in 19, similarly does not screen the α-face of the A–C rings of the steroid. Studies using Dreiding molecular models of macrocycles 1 and 19 indicate this is possible.
The literature results with macrocycle 5 indicate that the 7α-hydroxyl group, as expected, does not react and does not prevent the steroid derivative from binding to the CALB active site so that a successful ED-ROP can take place.18
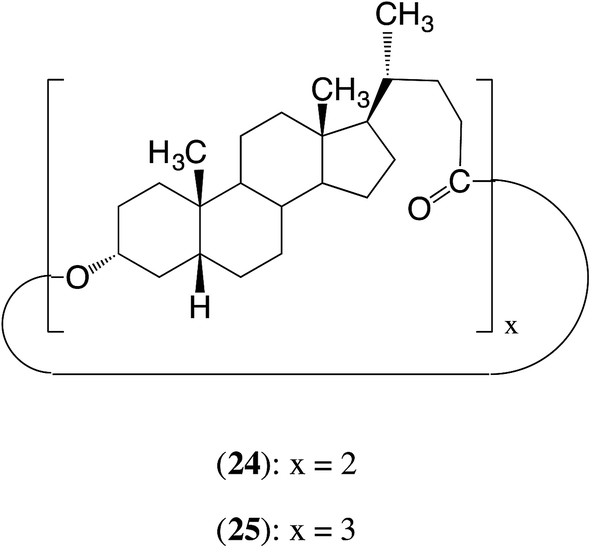
In our earlier work we showed that the cyclic dimer 24 and cyclic trimer 25 of lithocholic acid fail to polymerize with CALB.17 The present results suggest the reason for the failure may well be similar to that with 12 and 13, i.e. the appropriate part of the α-face of the steroid has difficulty accessing the active site, probably in this case because the α-faces of all the steroid units face inward towards the centres of the macrocycles.27 Another reason for the failure may be the relatively low reactivity of an ester group at the 24-position (see later in Section 2).
Selectivity studies using model bile acid derivatives
As discussed above, the bile acid-containing macrocycles that undergo ED-ROP successfully when treated with PS-CALB are 1, 2, 5, 11 and 19. This now prompts the question: what are the structures of the polymers produced? The ED-ROPs proceed via repeated TEs and to be able to predict the structures of the products it is necessary to know which of the various ester groups will react and, when they do react, their relative reactivity. This section discusses how this has been assessed by carrying out TEs with model bile acid derivatives.The diesters 26–28 and triester 29 were used in the model TEs: see the ESI† for details of their preparation. Between them these esters contain the three types of ester group present in the polymerizable macrocycles, i.e. 3α-esters, and esters at C24 and at C26 (see formula 29 for the numbering).


In general the TEs were carried out by treating the model compound with a 9-fold excess of an appropriate volatile unactivated ester: see Table 2. The reaction conditions used were essentially the same as those used in the ED-ROPs discussed above. Thus, the substrate in toluene at 70 °C was treated for 20 h with the volatile ester in the presence of 18 wt% of PS-CALB. At the end of the reaction period the polymer beads were filtered off and the filtrate evaporated to constant weight. This removed both the solvent and the volatile esters. The residue was then analyzed by 1H NMR spectroscopy. The volatile esters were chosen so that the reactions at the 3α- and/or 24-positions could be monitored easily: see ESI† for details. Since a 9-fold excess of volatile ester was used, if full equilibration is achieved, at the end of the TE at the 3α-position, for example, there would be 10 mol% of the original ester groups remaining and 90 mol% of the new ester groups. Retreatment of the product is expected to leave 1 mol% of the original bile acid ester groups and 99 mol% of the new ester groups. Typical 1H NMR traces of TEs (Table 2 entries 1, 4, 5 and 8) are included in the ESI.†
| Entry | Steroid seriesa | Starting materialb | Composition of starting materialc (%) | Reaction conditionse | Composition of reaction productc (%) | |||
|---|---|---|---|---|---|---|---|---|
| Ad | Bd | Added ester | Catalyst | Ad | Bd | |||
| a L = lithocholate series; D = deoxycholate series.b p = product from preceding entry.c As determined by 1H NMR spectroscopy. Errors in values estimated at ±5% of the values given.d A = 3α-acetate:3α-propionate B = –CO2CH3:–CO2C2H5.e Unless indicated otherwise reactions were carried out using ca. 0.70 mmol of steroid, 18 wt% of PS-CALB, with a 9 fold excess of the ester in 4.0 mL of toluene at 70 °C for 20 h.f Recrystallization of this final product from methanol gave a sample of methyl 3α-propionyloxy-cholanate (30) (78%) as white crystals, mp 137–138 °C.g Reaction time 7 days.h Reaction time 12 days.i Reaction time 3 h. |
||||||||
| 1 | L | 26 | 100:0 |
0:100 |
C2H5CO2C2H5 | CALB | 13:87 |
0:100 |
| 2 | L | p | 13:87 |
0:100 |
CH3CO2C2H5 | CALB | 94:6 |
0:100 |
| 3 | D | 27 | 100:0 |
100:0 |
C2H5CO2CH3 | CALB | 8:92 |
100:0 |
| 4 | D | p | 8:92 |
100:0 |
C2H5CO2CH3 | CALB | 10:90 |
100:0 |
| 5 | L | 28 | 100:0 |
100:0 |
C2H5CO2C2H5 | CALB | 10:90 |
95:5 |
| 6 | L | p | 10:90 |
95:5 |
C2H5CO2C2H5 | CALB | 5:95 |
90:10f |
| 7 | L | 28 | 100:0 |
100:0 |
CH3CO2C2H5 | CALB | 100:0 |
95:5 |
| 8 | L | 28 | 100:0 |
100:0 |
CH3CO2C2H5 | CALBg | 100:0 |
60:40 |
| 9 | L | 26 | 100:0 |
0:100 |
CH3CO2CH3 | CALB | 100:0 |
5:95 |
| 10 | L | 26 | 100:0 |
0:100 |
CH3CO2CH3 | CALBh | 100:0 |
70:30 |
| 11 | D | 27 | 100:0 |
100:0 |
CH3CO2C2H5 | CALB | 100:0 |
95:5 |
| 12 | L | 26 | 100:0 |
0:100 |
C2H5CO2CH3 | CALBi | 55:45 |
2.5:97.5 |
| 13 | L | 26 | 100:0 |
0:100 |
C2H5CO2CH3 | pTSA | 48:52 |
30:70 |
First various TEs were investigated using compounds 26–28. The results are summarized in Table 2. In the experiment summarized in entry 1, where diester 26 was treated with ethyl propionate, it was possible for the 3α-acetate to be converted into the propionate, but there could be no net reaction of the 24-position. It is evident from the results that the 3α-acetate transesterifies well under the reaction conditions. When the product was treated under the standard conditions with ethyl acetate, entry 2, the diester 24 was reformed. This indicates that the 3α-esters react efficiently under the present reaction conditions, and that the transesterification was fully reversible.
Similar reactions were carried out starting with the deoxycholic acid derivative 27. The result, see entry 3, further confirms that ester exchange at the 3α-position is facile and reversible (entry 4), and that the 12α-hydroxyl does not react.
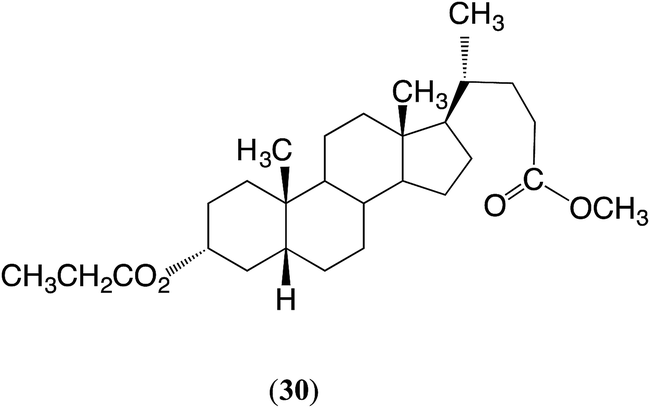
Diester 28 was then treated with ethyl propionate under the standard conditions: see entry 5. In this reaction TE might be observed at both the 3α-ester and the 24CO2CH3 ester. It is evident that the 3α-acetate reacted in high yield to give the 3α-propionate, but surprisingly the C24 ester also reacted, in 5% yield, to give the ethyl ester. The product was retreated with ethyl propionate as before (entry 6) in order to increase the content of the 3α-propionate, and the product was recrystallized twice from methanol. This gave a pure sample of methyl 3α-propionyloxycholanate (30) (78%), identified by the 1H NMR spectrum and confirmed by the melting point in comparison with the literature value.28
Attention was now focused on TE at the 24-position. To simplify the experiments volatile acetates were used which would result in no net reaction of the 3α-ester. The first experiment, summarized in entry 7, involved reacting compound 28 with ethyl acetate under the standard conditions. Under these conditions the C24 methyl ester was converted into the ethyl ester in ca. 5% yield. When the reaction time was increased to 7 days the conversion to the ethyl ester was 60%: see entry 8. In similar experiments with diester 26, see entries 9 and 10, the ethyl ester group in that substrate was converted into methyl ester 5% in 20 h and in 70% in 12 days. The bile acid derivative 27 reacted similarly: see entry 11.
To obtain a better estimate of the relative rates of reaction at the 3α-esters and the C24 esters, compound 26 was reacted with methyl propionate under the usual conditions except that the reaction time was reduced to just 3 h: see entry 12. In this time, 45% of the 3α-acetate was converted into the propionate, and 2.5% of the ethyl ester was converted into the methyl ester. Bearing in mind the maximum possible conversion at the 3-position is 90%, this suggests a relative rate of >20.
Finally diester 26 was treated with methyl propionate under the usual conditions except that the CALB was replaced by a catalytic amount of para-toluenesulfonic acid (pTSA). The results, see entry 13, indicate that both ester groups reacted significantly, with the 3α-acetate somewhat more reactive than the ethyl ester group.
Attention was then turned to the triester 29. The results are summarized in Table 3. TEs with this compound are obviously more complex than the previous TEs in that there are three different ester groups that might react and reaction at the 24-position detaches C25 and C26. The results of treating triester 29 with methyl propionate under the standard conditions are summarized in Table 3, entry 1. It is evident that, as in the previous experiments under similar conditions, the 3α-acetate transesterifies efficiently and that the ester at C24 also reacts, but only in ca. 5% yield. In addition C26 reacts efficiently. When the reaction time is reduced it is apparent that the rate of reaction at C26 is significantly less that the rate of reaction at the 3α position: compare entry 2. This suggests a factor of >2. When pTSA was used in place of CALB as the catalyst, all three ester sites reacted to a significant extent: see entry 3.
| Entry | Reaction conditionsa | Composition of reaction productb (%) | |||
|---|---|---|---|---|---|
| Time (h) | Catalyst | 3α-acetate:3α-propionate |
–CO2CH3 | C26-acetate:C26-propionate |
|
| a Unless indicated otherwise reactions were carried out using 0.70 mmol of steroid, 18 wt% of PS-CALB and a with a 9 fold excess of the ester in 4.0 mL of toluene at 70 °C for 20 h.b As determined by 1H NMR spectroscopy. Errors in values estimated at ±5% of the values given.c Values in parenthesis allows for loss of groups due to cleavage at C24. | |||||
| 1 | 20 | CALB | 10:90 |
5 | 3 (3):97 (92)c |
| 2 | 4 | CALB | 13:87 |
4 | 66 (63):34 (37) |
| 3 | 20 | pTSA | 8:92 |
41 | 41 (24):59 (35) |
In summary, the TE experiments indicate the decreasing order of the reactivity of the various ester groups is the 3α-OAc is >20 times more reactive than esters at the 24-position, and >2 times more reactive than the ester at the 26-position. This allows the structures of the products of the successful ED-ROPs to be predicted. This is discussed in Section 3.
The low reactivity of the ester group at the 24-position relative to that at the 3α-position is surprising given that the latter is separated from the rigid steroid core by three C–C. Also, given that the ester at the 3α-position is derived from a secondary alcohol whereas those at the 24-position are from methanol or ethanol. The fact that the ester group at the 26-position is more reactive than that at the 24-position is expected, as it is further removed from the steroid core, but the ester group at the 3α-position is still the most reactive. This suggests the steroid core fits particularly well onto the active site of CALB.
It should be noted that in the TE experiments reported in Table 2 entries 3, 4 and 11 there was, as expected, no evidence for reaction of the 12α-hydroxyl group.21–23
Structures of the various polymeric products
The key factor determining the structure of the polymers produced by the present ED-ROPs is the relatively low reactivity of the ester group at the 24-position. The ester groups at the 3- and 26-positions are significantly more reactive. Accordingly, the polymerizations generally proceed in two main stages. In the first stage reaction at the 24-position is relatively small, say, <5%. In the subsequent stage(s) all the ester groups react significantly. The above results suggest that the standard polymerization conditions used in this present work, i.e. 70 °C for 20 h, only allow stage 1 to be achieved. This will be especially the case at shorter reaction times (see Table 1, entries 4 and 7).Consider first the ED-ROP of macrocycle 1. In the first stage the polymeric product will be formed mainly (approximately 95%) by ring opening at the 3α-position, and only to a minor extent (approximately 5%) by ring opening at the 24-position. The former will generate the repeat unit 31, i.e. a structure in which the two types of ω-hydroxy acid formally present in 1 alternate. Reaction at the ester group at the 24-position will generate structure 32, in which the positions of the two ω-hydroxy acids are reversed. Thus there will be a small percentage of situations where two fatty acid units are next to each other and other places where two steroid units are next to each other. Over very long reaction periods both types of ester groups present in the polymer chains will react, stage 2, and eventually this will create a random order of the two types of ω-hydroxy acid units. The ED-ROPs of MCOs 11 and 19 will follow a similar pattern. Note that the structures of the polymers differ from those, for example 6 and 14, obtained by ED-ROMP.


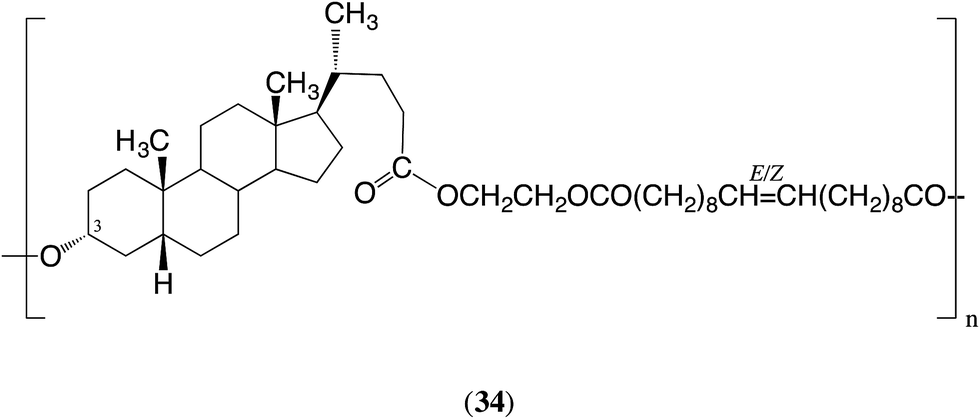
The situation with macrocycles 2 and 5, which each contain three ester groups, is potentially more complex. Consider first the polymerization of macrocycle 2. In the first stage of polymerization the unit 33 will behave as an α,ω-diol and the (symmetrical) E/Z-eicos-10-enedioic acid as a diacid. The ester groups linking these units will both react, with the 3α-position being somewhat more reactive (factor > 2). Thus, the polymeric product will have mainly structure 34 together with a smaller but significant amount of a similar structure with the ‘direction’ of the unsymmetrical diol unit 33 reversed. Over very long reaction periods all three types of ester groups present in the polymer chains will react and eventually this will create a scrambled order of the ω-hydroxy acid unit, the diacid and ethylene glycol units. In summary, in the first stage of the ED-ROP the product will have essentially the same structure as the product from the ED-ROMP of macrocycle 2. The only difference is that the structure of the product using CALB will have slightly more of the product with the diol unit 33 as shown in formula 34, whereas that from the ROMP is expected to have equal amounts of the two possible arrangements. The ED-ROP of macrocycle 5 will follow a similar pattern.
The ED-ROP of macrocycles 3 and 4 using PS-CALB has not been investigated. However, they would not be expected to proceed well as it is known that the ED-ROP of pentadecanolactone (17) affords polymer 18 with greatly reduced molecular weights in the presence of amide groups: see ESI† for details.
Conclusions
Macrocycles 1, 2 and 11 undergo ED-ROP successfully when treated with PS-CALB, but MCOs 12 and 13 fail to polymerize.Model TEs catalyzed by PS-CALB indicate that the decreasing order of ester group reactivity is 3α-OAc > 26 OAc ≫ 24-CO2R. The 12α-OH does not react.
Accordingly under the present polymerization conditions the polymeric product formed from the macrocycle 1 is expected to have mainly unit 31, i.e. that formed essentially by ring-opening at the 3α-position. This structure differs significantly from the structure of the polymers obtained by ED-ROMP of the same macrocycles. The latter, for example, contain a substantial fraction of E/Z-eicos-10-enedioic acid units. The ED-ROPs of MCOs 11 and 17 will follow a similar pattern.
Since macrocycle 2 contains three ester groups the course of the polymerization is more complex, but the product is expected to essentially have structure 34, with some of the diol unit 33 reversed. The polymerization of macrocycle 5 is expected to follow a similar course.
It is important to note that in all cases the initial structures obtained using PS-CALB differ, even if only in detail, from those obtained using ROMP.
Esterifications and TEs catalyzed by CALB using a large excess of appropriate volatile aliphatic esters are a convenient alternative to using the more expensive activated esters in the synthesis of esters using PS-CALB.21,22
Experimental
Materials and methods
These are as given in our earlier publication on PS-CALB.17Source of macrocycles
Macrocycles 1, 11, 12 and 13 were available from earlier published work.12 Macrocycle 2 was a kind gift from Professor X. X. Zhu, University of Montreal, Canada.ED-ROPs with PS-CALB
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1H NMR (500 MHz, CDCl3, δ, ppm) 5.30 (m, 2H, CH, E- and Z-isomers), 4.67 (m, 1H, 3β-CH), 3.98 (m, 2H, CH2OCO), 2.35–2.05 (m, 4H, 23-CH2 and CH2CO), 2.0–0.9 (m, 56H, various CH), 0.92 (s, 3H, C19), 0.90 (d, J = 10 Hz, 3H, C21 methyl), 0.67 (s, 3H, C18). SEC: Mn 19300; Mw 31700.O); 1H NMR (300 MHz, CDCl3, δ, ppm) 5.37 (m, 2H, CH, E- and Z-isomers), 4.75 (m, 1H, 3β-CH), 4.02 (m, 4H, OCH2CH2O), 2.35–2.05 (m, 6H, 23CH2 and 2CH2CO), 2.0–0.9 (m, 54H, various CH), 0.92 (s, 3H, C19), 0.90 (d, J = 10 Hz, 3H, C21 methyl), 0.64 (s, 3H, C18). SEC: Mn 20500; Mw 46500.O); 1H NMR (500 MHz, CDCl3, δ, ppm) 5.33 (m, 2H, CH, E- and Z-isomers), 4.67 (m, 1H, 3β-CH), 3.98 (m, 2H, CH2OCO), 3.90 (m, 1H, 12β-H), 2.35–2.05 (m, 4H, 23-CH2 and CH2CO), 2.0–0.9 (m, 54H, various CH), 0.90 (d, J = 10 Hz, 3H, C21 methyl), 0.83 (s, 3H, C19), 0.62 (s, 3H, C18). SEC: Mn 15300; Mw 30700.
O); 1H NMR (500 MHz, CDCl3, δ, ppm) 5.30 (m, 2H, CH, E- and Z-isomers), 4.67 (m, 1H, 3β-CH), 3.98 (m, 2H, CH2OCO), 2.35–2.05 (m, 4H, 23-CH2 and CH2CO), 2.0–0.9 (m, 56H, various CH), 0.92 (s, 3H, C19), 0.90 (d, J = 10 Hz, 3H, C21 methyl), 0.67 (s, 3H, C18). SEC: Mn 19300; Mw 31700.O); 1H NMR (300 MHz, CDCl3, δ, ppm) 5.37 (m, 2H, CH, E- and Z-isomers), 4.75 (m, 1H, 3β-CH), 4.02 (m, 4H, OCH2CH2O), 2.35–2.05 (m, 6H, 23CH2 and 2CH2CO), 2.0–0.9 (m, 54H, various CH), 0.92 (s, 3H, C19), 0.90 (d, J = 10 Hz, 3H, C21 methyl), 0.64 (s, 3H, C18). SEC: Mn 20500; Mw 46500.O); 1H NMR (500 MHz, CDCl3, δ, ppm) 5.33 (m, 2H, CH, E- and Z-isomers), 4.67 (m, 1H, 3β-CH), 3.98 (m, 2H, CH2OCO), 3.90 (m, 1H, 12β-H), 2.35–2.05 (m, 4H, 23-CH2 and CH2CO), 2.0–0.9 (m, 54H, various CH), 0.90 (d, J = 10 Hz, 3H, C21 methyl), 0.83 (s, 3H, C19), 0.62 (s, 3H, C18). SEC: Mn 15300; Mw 30700.Preparation of model bile acid derivatives
The preparations of compounds 26–29 are described in the ESI.†Entry 3. Diester 27 (310 mg, 0.69 mmol) in toluene (4.0 mL) at 70 °C was treated for 20 h with methyl propionate (546 mg, 6.21 mmol) and PS-CALB (58 mg) under an atmosphere of dry nitrogen. The beads were then filtered off and the mixture evaporated to leave a residue of constant weight (97% mass recovered). The 1H NMR spectrum was recorded. This indicated the product had the composition reported in Table 1.
Entries 5 and 6. The product from the reaction summarized in entry 5 was retreated (entry 6) under the same reaction conditions. Recrystallization of the final product from methanol gave methyl 3α-propionyloxycholanate (30) (78%) as white crystals, mp 137–138 °C (lit.,28 138–140 °C); 1H NMR (300 MHz, CDCl3, δ, ppm) 4.72 (m, 1H, 3β-CH), 3.66 (s, 3H, OCH3), 2.40–2.10 (m, 4H, 23-CH2 and CH2CO), 2.0–1.0 (m, 26H, various CH), 0.92 (s, 3H, C19), 0.90 (d, J = 10 Hz, 3H, C21 methyl), 0.63 (s, 3H, C18).
Acknowledgements
We thank Professor X. X. Zhu, University of Montreal, for a gift of macrocycle 2, and Drs John J. Morrison and Daniel J. Tate for assistance recording some of the spectra.References
- P. Hodge, Chem. Rev., 2014, 114, 2278 CrossRef PubMed.
- See, for example: (a) P. Hodge and H. M. Colquhoun, Polym. Adv. Technol., 2005, 6, 84 CrossRef; (b) D. J. Brunelle, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1151 CrossRef CAS; (c) Z. Xue and M. F. Mayer, Soft Matter, 2009, 5, 4600 RSC; (d) S. Strandman, J. E. Gautrot and X. X. Zhu, Polym. Chem., 2011, 2, 791 RSC.
- (a) H. Jacobson and W. H. Stockmayer, J. Chem. Phys., 1950, 18, 1600 CrossRef CAS; (b) H. Jacobson, C. O. Beckmann and W. H. Stockmayer, J. Chem. Phys., 1950, 18, 1607 CrossRef CAS; (c) E. Thorn-Csanàyi and K. Ruhland, Macromol. Chem. Phys., 1999, 200, 1662 CrossRef; (d) A. Hejl, O. A. Scherman and R. H. Grubbs, Macromolecules, 2005, 38, 7214 CrossRef CAS; (e) E. A. Ofstead and N. Calderon, Makromol. Chem., 1972, 154, 21 CrossRef CAS; (f) H. Höcker, W. Reimann, L. Reif and K. Riebel, J. Mol. Catal., 1980, 8, 191 CrossRef; (g) G. Ercolani, L. Mandolini, P. Mencareli and S. Roelens, J. Am. Chem. Soc., 1993, 115, 3901 CrossRef CAS; (h) G. Ercolani, L. Mandolini and P. Mencareli, J. Chem. Soc., Perkin Trans. 2, 1990, 747 RSC; (i) A. Dalla Cort, G. Ercolani, L. Mandolini and P. Mencareli, J. Chem. Soc., Chem. Commun., 1993, 538 RSC.
- C. L. Ruddick, P. Hodge, Y. Zhuo, R. L. Beddoes and M. Helliwell, J. Mater. Chem., 1999, 9, 2399 RSC.
- J. E. Gautrot and X. X. Zhu, Angew. Chem., Int. Ed., 2006, 45, 6872 CrossRef CAS PubMed.
- J. E. Gautrot and X. X. Zhu, Macromolecules, 2009, 42, 7324 CrossRef CAS.
- (a) P. Hodge, Polym. Adv. Technol., 2015, 26, 797 CrossRef CAS; (b) S. Matsumura, Macromol. Biosci., 2002, 2, 105–126 CrossRef CAS.
- (a) P. Hodge, React. Funct. Polym., 2014, 80, 21 CrossRef CAS; (b) Y. Osanai, K. Toshima and S. Matsumura, Sci. Technol. Adv. Mater., 2006, 7, 202 CrossRef CAS.
- A. C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466 CrossRef CAS PubMed.
- (a) X. X. Zhu and M. Nichifor, Acc. Chem. Res., 2002, 35, 539 CrossRef CAS PubMed; (b) J. E. Gautrot and X. X. Zhu, J. Biomater. Sci., Polym. Ed., 2006, 17, 1123 CrossRef CAS.
- (a) A. Ivanysenko, S. Strandman and X. X. Zhu, Polym. Chem., 2012, 3, 1962 RSC; (b) J. E. Gautrot and X. X. Zhu, J. Mater. Chem., 2009, 19, 5705 RSC; (c) J. K. Denike and X. X. Zhu, Macromol. Rapid Commun., 1994, 15, 459 CrossRef CAS.
- P. Hodge and A. B. Chakiri, Polym. Chem., 2015, 6, 7274 RSC.
- (a) J. E. Gautrot and X. X. Zhu, Chem. Commun., 2008, 1674 RSC; (b) H. Thérien-Aubin, J. E. Gautrot, Y. Shao, J. Zhang and X. X. Zhu, Polymer, 2010, 51, 22 CrossRef.
- (a) S. Matsumura, Adv. Polym. Sci., 2006, 194, 95 CrossRef CAS; (b) R. A. Gross, A. Kumar and B. Kalra, Chem. Rev., 2001, 101, 2097 CrossRef CAS PubMed; (c) L. van der Mee, F. Heimich, R. de Bruijn, J. A. J. M. Vekemans, A. R. A. Palmans and E. W. Meijer, Macromolecules, 2006, 39, 5021 CrossRef CAS.
- See, for example: (a) M. L. Focarete, M. Scandola, A. Kumar and R. A. Gross, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 1721 CrossRef CAS; (b) I. van der Meulen, Y. Li, R. Deumens, E. A. J. Joosten, C. E. Koning and A. Heise, Biomacromolecules, 2011, 12, 837 CrossRef CAS PubMed.
- Y. Poojari, J. S. Beemat and S. J. Clarson, Polym. Bull., 2013, 70, 1543 CrossRef CAS.
- B. Manzini, P. Hodge and A. Ben-Haida, Polym. Chem., 2010, 1, 339 RSC.
- S. Strandman, I.-H. Tsai, R. Lortie and X. X. Zhu, Polym. Chem., 2013, 4, 4312 RSC.
- (a) J. Uppenberg, M. T. Hansen, S. Patkar and T. A. Jones, Structure, 1994, 2, 293 CrossRef CAS PubMed; (b) J. Uppenberg, N. Öhrner, M. Norin, K. Hult, G. J. Kleywegt, S. Patkar, V. Waagen, T. Anthonsen and T. A. Jones, Biochemistry, 1995, 34, 16838 CrossRef CAS PubMed.
- See, for example, (a) A. Ferscht, in Enzyme Structure and Mechanism, W. H. Freeman and Co, Reading, 1977, pp. 302–312 Search PubMed; (b) D. M. Blow, J. J. Birktoft and B. S. Hartley, Nature, 1969, 221, 337 CrossRef CAS PubMed.
- A. Bertinotti, G. Carrea, G. Ottolina and S. Riva, Tetrahedron, 1994, 50, 13165 CrossRef CAS.
- T. Sugai, M. Takzawa, M. Bakke, Y. Ohtsuka and M. Ohta, Biosci., Biotechnol., Biochem., 1996, 60, 2059 CrossRef CAS PubMed.
- O. Noll and H. Ritter, Macromol. Rapid Commun., 1996, 17, 553 CrossRef CAS.
- P. G. Quintana and A. Baldessari, Steroids, 2009, 74, 1007 CrossRef CAS PubMed.
- (a) A. Baldessari, A. C. Bruttomesso and E. C. Gros, Helv. Chim. Acta, 1996, 79, 999 CrossRef CAS; (b) A. C. Bruttomesso and A. Baldessari, J. Mol. Catal. B: Enzym., 2014, 29, 149 CrossRef.
- R. G. Gilbert, M. Hess, A. D. Jenkins, R. G. Jones, P. Kratochvil and R. F. T. Stepto, Pure Appl. Chem., 2009, 81, 351 CAS.
- (a) P. A. Brady, R. P. Bonar-Law, S. J. Rowan, C. J. Suckling and J. K. M. Sanders, Chem. Commun., 1996, 319 RSC; (b) R. P. Bonar-Law and J. K. M. Sanders, Tetrahedron Lett., 1992, 33, 2071 CrossRef CAS; (c) Y. Li and J. R. Dias, Synthesis, 1997, 425 CrossRef CAS.
- F. Hodosan and A. Pop-Gogan, Rev. Roum. Chim., 1969, 14, 1441 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra17954h |
| This journal is © The Royal Society of Chemistry 2015 |