
Low hazard small-scale synthesis and chemical analysis of high purity nitroglycerine (NG)
Alessandro E. Contini,
Nathan Flood *,
Daniel McAteer,
Nathalie Mai and
Jacqueline Akhavan
*,
Daniel McAteer,
Nathalie Mai and
Jacqueline Akhavan
Centre for Defence Chemistry, Cranfield University, Defence Academy of the United Kingdom, Shrivenham, SN6 8LA, UK. E-mail: n.flood@cranfield.ac.uk
First published on 6th October 2015
Abstract
A previously reported two-phase (99.5% fuming nitric acid/dichloromethane) batch method to prepare high purity 1,2,3-propanetriyl trinitrate (nitroglycerine, NG) was evaluated, simplified and adapted specifically for low hazard small-scale synthesis. The purity of the product, as determined by NMR spectroscopy, HPLC and IR spectroscopic analysis was found to be greater than 99%. The quick synthetic method is highly recommended when small amounts of highly pure NG are required for analytical and related purposes in the absence of a microreactor.
Introduction
As a key component of double base propellants and a potent vasodilator2–5 nitroglycerine (1,2,3-propanetriyl trinitrate or NG) is a compound of common interest to both energetic materials and pharmaceutical scientists. Due to its effectiveness as an energetic additive and as a medicine, it has been produced on the industrial scale for well over a century.6–8 Despite this, the technical literature is still scant of simple synthetic batch methods9–11 to prepare NG on very small-scales (less than 1 g) as may be required from time to time for analytical purposes by energetic materials and pharmaceutical research laboratories, which have no access to microreactor technology. On such small scales, traditional mixed-acid nitration of neat liquid glycerol can still be hazardous because of the sharp exotherm generated by the addition of the polyol to the chilled mixed acids. In the case of NG, the reaction temperature and the addition rate of glycerol to the nitration medium must be carefully controlled in order to avoid fume-off and/or potential explosion due to hydrolytic processes.12 Using mixed acid nitration also limits the purity and published work does not report the potentially achievable purity.13 Due to these drawbacks, synthetic chemists working in licensed laboratories may be discouraged or even forbidden from attempting to synthesise even minute quantities of neat NG without prior training in nitration theory and practice. This need not be the case if a proven and suitable small-scale synthetic protocol is followed.Historically there have been many reported accidents during the synthesis, storage, handling and processing of NG which have caused numerous injuries and fatalities.14 More recently reported explosive incidents have occurred during the processing of NG.15,16 The large-scale manufacture of NG is highly specialised and heavily regulated due to safety and security concerns.17 Currently in the UK there is only one large-scale manufacturer of NG.† This creates a problem for chemists in acquiring NG of high purity for research and development.
In recent literature the characterisation and material properties of NG have not been thoroughly researched due to the issues previously stated relating to purchasing and small-scale production. The majority of the research into characterising NG is related to propellant research,18,19 which mainly focuses on the properties of this nitrate ester as an energetic plasticiser.20,21 Some published infrared spectra exist but most of these are historic and lack the sensitivity of modern instrumentation.13,22,23 Historically many analytical methods, both quantitative and qualitative have been used,24 but the most common technique for quantitative analysis is HPLC.25,26 There are, in fact, multiple HPLC methods and procedures that have been developed by the pharmaceutical27,28 and forensic fields29,30 however commercial NG is expensive and is only available in small quantities (typically tens of milligrams). If a safer method for small-scale synthesis (∼1 g) was developed, it would reduce the time and costs of routine analysis.
This paper presents a new method for the synthesis of NG for small-scale batch processes, which has a purity of 99% and is less hazardous to prepare than the previously reported synthetic routes.9–11
Warning: As with other low molecular weight nitrate esters, dry, pure NG is extremely sensitive to impact and friction. Despite the relatively low hazard of the suggested synthetic procedure, both synthesis and isolation methods described herein should only be attempted by competent chemists working in a licenced explosives laboratory. During isolation of the neat liquid product, Kevlar gloves and a face shield must be worn. Scaling up the isolation of neat NG above 40 mg by rotary evaporation behind a blast screen, could be dangerous due to potential initiation of the liquid NG by entrained gas bubbles, due to the formation of hot spots through adiabatic collapse of the gas bubbles.31
Experimental
Synthetic procedure
99.5% fuming nitric acid (1.1 ml, 26.3 mmol), was added via a 5 ml pressure-equalising funnel to magnetically stirred 98+% sulphuric acid (1.1 ml) in a 10 ml round bottomed flask immersed in an ice-bath, thus ensuring that the temperature of the mixture did not rise above 10 °C. Dichloromethane (DCM) (8 ml) was then added in a single portion to the mixed acid and the mixture was left stirring and cooled below 5 °C. Glycerol (500 mg, 5.4 mmol) was manually added drop-wise, with the aid of a wide-nozzle Pasteur pipette, to the rapidly-stirred acid/DCM mixture at such a rate as to maintain the temperature below 15 °C. As glycerol is not soluble in DCM, the drops initially swirled in the rapidly stirred mixture and gradually decreased in size as the glycerol reacted. A concomitant increase in temperature was observed. After about 10 min the addition was complete, the mixture was stirred for a further 50 min, after which time the mixture lost its opalescent appearance and became clear. It was then transferred to a 20 ml separating funnel and the mixed acid lower layer was run off and discarded. The organic layer was then washed as follows: water (1 × 3 ml), 10% aqueous sodium carbonate (Na2CO3) (3 × 2 ml), 10% aqueous urea (1 × 3 ml), water (3 × 2 ml) and finally brine (1 × 2 ml). The washed organic phase was dried over magnesium sulphate (MgSO4) and filtered to yield a solution of 1,2,3-propanetriyl trinitrate (NG) in DCM.The solvent from the DCM solution (6.9 wt% NG) was removed from a 0.5 ml volumetric aliquot. This was carried out with a standard rotary evaporator located behind a blast screen situated in a ventilated fume cupboard. A Teflon sleeve was placed between the male and female ground glass joints of the pre-tared recovery flask and the rotary evaporator stem. Working with Kevlar gloves behind the blast screen, the flask containing small droplets of neat NG (∼35 mg, as a clear, colourless mobile oil) was weighed and immediately re-dissolved in a deuterated solvent for NMR analysis. The weighed mass of the droplet was used to calculate the yield of reaction and also to work out the exact concentration of the DCM solution.
Yield: 1.05 g (85%). NMR (400 MHz, acetone-d6) 1H: 4.95–5.00 ppm (dd, 2.02H, C1 Ha and C3 Ha, see Fig. 1), 5.14–5.18 ppm (dd, 2.04H, C1 Hb and C3 Hb) and 5.90–5.94 ppm (m, 1.00H, C2 methine proton); 13C: 71 ppm (C1 and C3) and 78 ppm (C2); 14N: 290 ppm (C2 ONO2) and 292 ppm (C1 and C3 ONO2).
 | ||
| Fig. 1 1H NMR spectrum (in acetone-d6) of NG. | ||
Disposal
Small surplus quantities of neat NG may be safely destroyed by re-dissolving the neat liquid in acetone, or other low toxicity flammable solvent, for controlled burning in a ventilated fume cupboard. Ensure that the NG is under no confinement if burning, as a detonation would be expected even with minimal sample.NMR spectroscopy
All NMR spectra were recorded at 300 K on a Bruker Avance III ‘NanoBay’ 400 MHz spectrometer fitted with a broadband BBFO probe. The chemical shifts are quoted in parts per million (ppm) with reference tetramethylsilane (TMS) for 1H and 13C spectra recorded in acetone-d6.ATR-FT-IR spectroscopy
IR spectra were recorded on a Bruker ALPHA using Opus 7.2 software. The resolution was 1 cm−1 at 128 scans. A few small drops of the mother liquor were deposited on a glass slide, the DCM was allowed to evaporate off, and the slide was then placed on a diamond attenuated total reflection (ATR) accessary and analysed.HPLC
Reverse phase HPLC analyses were performed using a Waters Alliance 2695 HPLC and a Waters 996 PDA detector. The column was a 100 × 4.6 mm Kinetex 2.6 μm C18 100 Å, sourced from Phenonemex UK, and was set to 30 °C. The mobile phase was 50% acetonitrile (ACN)/H2O at a flow rate of 1.5 ml min−1. The injection volume was 10 μl. ISO 17025 certified NG standards were purchased from AccuStandard Laboratory (New Haven, USA). HPLC grade ACN was obtained from Fisher Scientific (Loughborough, Leicestershire, UK). High purity water was prepared by using Millipore Milli Q plus purification system. Four calibration standards prepared in ACN ranging from 20 to 50 ppm were injected in duplicate. The DCM solution (0.5 ml) of the synthetic NG sample was evaporated and 10 ml of ACN was added to the sample to prepare the stock solution. The solution was diluted by 100 in ACN and analysed in duplicate by HPLC. The equation of the resulting calibration curve was used to calculate the actual amount of NG contained in the volumetric aliquot.Results and discussion
In our search for a relatively low-hazard batch method to synthesise small quantities of dry, analytically pure NG, we have evaluated and optimized an existing literature method1 with a view to synthesise milligram to gram quantities of pure NG. Whilst the original procedure was developed to prepare batch quantities up to 450 g of various nitrate esters free of less stable nitrite (ester) impurities, and which relied on a redox potentiometric probe to regulate the glycerol addition rate, we have adapted the method to prepare much smaller amounts of product down to 250 mg, without using microreactors and without potentiometric control.Our method relies on two-phase (mixed acid and DCM) nitration of glycerol, whereby an inert and dense chlorinated solvent acts as a temperature-moderating heat sink during the reaction, removing the nitrate ester as it forms from the nitration medium, maximising the reaction yield by minimizing oxidative side reactions, and most importantly, de-sensitising the nitrate ester for safer handling. This is in contrast to traditional methods of NG synthesis where the product is stored as an aqueous emulsion which poses an explosive hazard upon separation.6 The product is typically obtained as a solution in dichloromethane, which is neutralised with aqueous bicarbonate and then repeatedly washed with water to remove less nitrated and/or oxidative side products. The final dry solution is non-flammable, and may be safely stored in a dark cupboard at room temperature for several months without any explosive risk.
From a licensing, storage and transport point of view this method also has the advantage that the NG is immediately dissolved in DCM. According to the United Nations Economic Commission for Europe's (UNECE) European Agreement concerning the International Carriage of Dangerous Goods by Road (ADR 2015)32 the desensitised non-flammable NG solution can be given a UN no. 3357 and is class 3D based on the ADR 2015.32
During preparation of the mixed acid on scales up to 1.0 g, we found that drop-wise addition of 99.5% nitric acid to chilled (0 °C) 98% sulphuric acid is more practical than adding sulphuric acid to chilled nitric acid, as a consequence of the viscosity of the former which prevents it from dripping freely through the narrow channel of a small dropping funnel. Although the reverse addition order is not recommended on larger scales, the acid dilution exotherm was negligible when a simple ice-water bath was used to chill the magnetically stirred sulphuric acid.
Gravimetric,‡ HPLC and 1H/13C/14N NMR spectroscopic analyses of the product (Fig. 1–5) confirmed that this method gives NG of very high purity (>99%) and in excellent yield. The analysis did not reveal the presence of unreacted glycerol, less nitrated by-products (1,2- and/or 1,3-dinitroglycerine) or nitrite esters, confirming that potentiometric control during glycerol addition is not required to obtain analytical purity on very small-scales (Table 1).
 | ||
| Fig. 2 13C NMR spectrum (in acetone-d6) of NG. | ||
 | ||
| Fig. 3 14N NMR spectrum (in acetone-d6) of NG. | ||
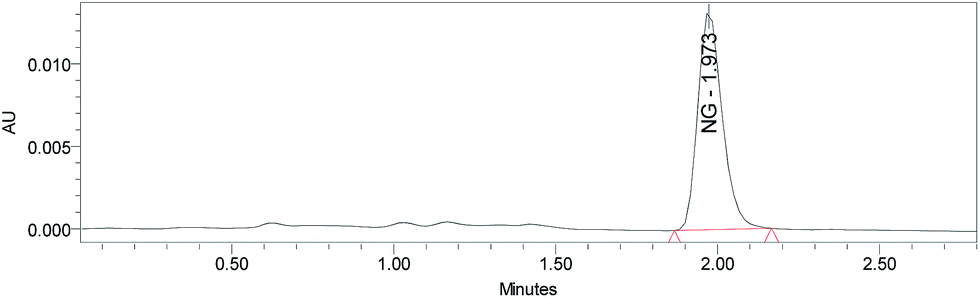 | ||
| Fig. 4 HPLC chromatogram of synthetic NG extracted at 254 nm. | ||
 | ||
| Fig. 5 Infrared spectrum of NG. | ||
| Analytical method | NG mass contained in 0.5 ml volumetric aliquot of stock DCM solution | Yield | Concentration/purity |
|---|---|---|---|
| Theoretical | 35.0 mg | — | 35.0 mg |
| Gravimetric | 34.4 mg | 99%+ | — |
| HPLC | 34.5 mg | — | 34.5 mg |
| NMR | — | 99%+ | — |
The 400 MHz 1H NMR spectrum of our product indicates purity greater than 99% (only 13C satellites are visible when the intensity of the spectrum is increased). The unsymmetrical signal shown in Fig. 1 can be assigned when considering the symmetrical nature of the NG molecule. Pro-chirality at C2 renders the protons of both methylene groups non-equivalent and this results in a more complex spectrum than would be expected from a simple AB spin system. The 13C spectrum (Fig. 2) displays only two product signals indicative of the C2 and the isosynchronous C1 and C3. The analysis of the product by 14N NMR spectroscopy (Fig. 3) (previously unreported) shows C1 and C3 nitrate esters as a broad peak at 292 ppm, which is approximately double the intensity of a very close peak resonating at 290 ppm, which was attributed to the nitrate ester at C2.
Fig. 4 depicts the HPLC chromatogram of the NG sample, showing the absence of impurities. This is further supported by Fig. 5, which displays the ATR-FT-IR spectrum of the NG sample. Some key absorption frequencies observed in the NG IR spectrum are presented in Table 2.
| Observed peaks (cm−1) | Functional group |
|---|---|
| 3024 | C–H stretch |
| 2966 | C–H stretch |
| 2916 | C–H stretch |
| 1631 | –NO2 asymmetric stretch |
| 1457 | –NO2 asymmetric stretch |
| 1428 | C–H bend/rock |
| 1376 | C–H bend/rock |
| 1293 | –NO2 symmetric stretch |
| 1264 | –NO2 symmetric stretch |
| 1087 | C–O stretch |
| 1052 | C–O stretch |
| 1007 | N–O linkage π bond stretching |
| 899 | C–H bend |
| 820 | –NO2 wag |
| 749 | C–H bend |
| 700 | C–H bend |
| 668 | C–H bend |
| 591 | C–H bend |
Fig. 6 shows NMR spectroscopic analysis of a two-year-old solution containing approx. 20 wt% of our synthesised NG. The NG did not reveal any degradation of the product after this time; this was also confirmed by HPLC (Fig. 7) and ATR-FTIR-IR.§
 | ||
| Fig. 6 1H NMR spectrum (in acetone-d6) of NG showing no degradation over two years (red – date of synthesis and blue – aged two years). | ||
 | ||
| Fig. 7 HPLC chromatogram of synthetic NG extracted at 254 nm showing no degradation after 2 years. | ||
If required, small quantities of up to 50 mg of neat product may be isolated as a colourless mobile oil by rotary evaporation of small volumetric aliquots of the dichloromethane mother liquor. Because of the very high purity and the total absence of water, the neat liquid NG obtained by this method has extremely high sensitiveness, particularly to impact. A small droplet deposited on a metal anvil can be readily initiated by application of a gentle hammer blow (Fig. 8), confirming the absence of less nitrated products and water that would greatly de-sensitise NG towards impact.
 | ||
| Fig. 8 NG detonating under a hammer blow. | ||
Conclusion
A previously published two-phase nitration method was successfully simplified and adapted to the small-scale (up to 1.0 g) synthesis of NG for analytical purposes. The product was obtained in excellent yield and high purity, as determined by gravimetric analysis, HPLC, ATR-FT-IR and NMR spectroscopy. Given the simplicity, relatively low hazard and cost involved, this synthetic method is highly recommended, whenever small quantities of highly pure NG (and possibly other nitrate esters) are required for analytical and related purposes in the absence of a microreactor.Acknowledgements
Thanks to the British Broadcasting Corporation (BBC) for assisting in photographing the ignition by impact of NG (Image hosted at BBC Motion Gallery). Thanks to Gavin Dunhill (Cranfield University) for his advice on hazard classifications.Notes and references
- C. Marken, C. Kristofferson, M. Roland, A. Manzara and M. Barnes, Synthesis, 1977, 484–485 CrossRef CAS.
- M. Pröbster and R. H. Schmucker, Acta Astronaut., 1986, 13, 599–605 CrossRef.
- L. Ignarro, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7816–7817 CrossRef CAS PubMed.
- J. R. Parratt, J. Pharm. Pharmacol., 1979, 31, 801–809 CrossRef CAS PubMed.
- G. Thatcher, Chem. Soc. Rev., 1998, 27, 331–337 RSC.
- A. S. Gordon and P. C. W. Art, Manufacture of explosives, US Pat., US2140897A, 1938.
- F. Nathan, J. Thomson and W. Rintoul, Apparatus for the manufacture of nitroglycerin, Report US702325A, Google Patents, US, 1902.
- C. A. Woodbury and W. C. Holmes, Ind. Eng. Chem., 1935, 27, 632–637 CrossRef CAS.
- S. Braune, S. Steinhofer, P. Poechlauer, R. W. E. G. Reintjens, N. T. W. Linssen and M. Thathagar, Formation of nitrate esters in microreactors and millireactors using a continuous product extraction in a turbulent flow regime, US Pat., US 12/808,829, 2011.
- J. Hogan, Nature, 2006, 442, 352 Search PubMed.
- A. Turley, Chemistry World, RSC, Cambridge, 2012 Search PubMed.
- E. Camera, G. Modena and B. Zotti, Propellants, Explos., Pyrotech., 1982, 7, 66–69 CrossRef CAS PubMed.
- R. Schneider and M. Hanson, An Infrared Spectroscopic Study of the Vapor Adsorption of Nitroglycerin (NG) on Chrysotile Asbestos, Colorado, 2000 Search PubMed.
- UK Health and Safety Executive, Explosives Incidents Database Advisory Service, http://webcommunities.hse.gov.uk/connect.ti/explosives/groupHome, accessed 7/1/2015.
- Munitions Safety Information Analysis Center, Lettre du MSIAC, NATO, 2nd Quarter 2011 Search PubMed.
- Munitions Safety Information Analysis Center, Lettre du MSIAC, NATO, 4th Quarter 2008 Search PubMed.
- UK Health and Safety Executive, Explosives Regulations 2014, http://www.hse.gov.uk/explosives/new-regulations.htm, accessed 3/8/2015.
- S. Heng, T. Pan, Y. Kong and Z. Liu, Propellants, Explos., Pyrotech., 1991, 16, 31–35 CrossRef CAS PubMed.
- F. Volk, G. Wunsch and D. Müller, Propellants, Explos., Pyrotech., 1982, 7, 53–57 CrossRef CAS PubMed.
- Y. M. Lotmentsev and D. V. Pleshakov, Propellants, Explos., Pyrotech., 1997, 22, 203–206 CrossRef CAS PubMed.
- D. A. Winkler, Propellants, Explos., Pyrotech., 1985, 10, 43–46 CrossRef CAS PubMed.
- F. Pristera, Anal. Chem., 1953, 25, 844–856 CrossRef CAS.
- F. Pristera, M. Halik, A. Castelli and W. Fredericks, Anal. Chem., 1960, 32, 495–508 CrossRef CAS.
- V. Y. Grigor'eva, O. A. Efremenko and Y. Y. Kharitonov, Pharm. Chem. J., 2004, 38, 562–568 CrossRef.
- R. Martel, A. Bellavance-Godin, R. Lévesque and S. Côté, Chromatographia, 2010, 71, 285–289 CAS.
- X. Xu, M. Koeberg, C. Kuijpers and E. Kok, Sci. Justice, 2014, 3–21 CrossRef PubMed.
- L. Gelber, J. Pharm. Sci., 1980, 69, 1084–1086 CrossRef CAS PubMed.
- A. Halasz, S. Thiboutot, G. Ampleman and J. Hawari, Chemosphere, 2010, 79, 228–232 CrossRef CAS PubMed.
- J. B. F. Lloyd, J. Forensic Sci. Soc., 1986, 26, 341–348 CrossRef CAS.
- F. M. Green, T. L. Salter, P. Stokes, I. S. Gilmore and G. O'Connor, Surf. Interface Anal., 2010, 42, 347–357 CrossRef CAS PubMed.
- F. P. Bowden and A. D. Yoffe, Initiation and Growth of Explosion in Liquids and Solids, Cambridge-Hitachi, 1985 Search PubMed.
- United Nations Economic Commission for Europe, European Agreement concerning the International Carriage of Dangerous Goods by Road (ADR 2015) 2015, 141.
Footnotes |
| † Roxel (UK Rocket Motors) Ltd. |
| ‡ Gravimetric analysis is the physical weighing of the NG residue after evaporating the DCM solvent from a 0.5 ml aliquot of stock solution. |
| § Results not shown, as there was no change from the spectra shown in Fig. 5. |
| This journal is © The Royal Society of Chemistry 2015 |