
Effects of functional groups of graphene oxide on the electrochemical performance of lithium-ion batteries†
Zhengwei Xieab,
Zuolong Yua,
Weifeng Fana,
Gongchang Penga and
Meizhen Qu*a
aChengdu Institute of Organic Chemistry, Chinese Academy of Sciences, Chengdu 610041, PR China. E-mail: mzhqu@cioc.ac.cn; xiezhengwei116@hotmail.com; Fax: +86 28 85215069; Tel: +86 28 85228839
bGraduate University of Chinese Academy of Sciences, Beijing 100039, PR China
First published on 7th October 2015
Abstract
Graphene oxide (GO) with different ratios of functional groups are prepared via low temperature direct thermal reduction technology and re-oxidization by nitric acid, respectively. The structural, elemental, and oxygen-containing functional group compositions and electrochemical behaviors of the prepared GO are characterized using Fourier infrared spectroscopy, X-ray diffraction, thermogravimetric analysis, Raman spectroscopy, and X-ray photoelectron spectroscopy, as well as charge/discharge curves and electrochemical impedance spectra measurements. Compared with graphite and graphene, the enhanced reversible capacity of GO is attributed to the oxygen-containing functional groups and the improved capacities are attributed predominantly to carbonyl and carboxyl groups. Besides, labile oxygen functionalities, such as epoxy groups, have a negative effect on the electrochemical properties, which lower the initial coulomb efficiency of graphene oxide anode materials. These findings may be beneficial to the material design of graphene-based anode materials with a high energy density.
1. Introduction
Graphite, which is the most common anode material for current commercial Li ion batteries (LIBs), cannot fulfill the requirements of high-energy applications because of its limited specific capacity with a theoretical value of 372 mA h g−1.1–3 Different types of anode materials with high specific capacities have been proposed for LIBs. Among these anode materials, carbonaceous materials have gained attention due to their low price and plentiful supply. In particular, graphene,4–9 with double the storage capacity of common graphite in theory, has been suggested as an attractive candidate for potential applications in electrochemical energy storage. However, higher costs, irreversible aggregation and hazardous or toxic reducing agents (NaBH4, hydrazine and formaldehyde) still seriously hinder its large-scale application.As the precursor of graphene, graphene oxide (GO) has been widely used in supercapacitors,10–12 fuel cells13 and Li-ion batteries.14–16 GO possesses a higher capacity and an outstanding cycling performance, and can easily form self-assembly membranes instead of polymer binders because of the numerous functional groups, such as hydroxyl, epoxy, carbonyl, carboxyl and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds. However, not all of these functional groups can play a positive role in the electrochemical performance; some of them have negative effects, such as a higher irreversible Li ion consumption.
C double bonds. However, not all of these functional groups can play a positive role in the electrochemical performance; some of them have negative effects, such as a higher irreversible Li ion consumption.
So far, there have been no final conclusions about the influence of different functional groups of GO on the electrochemical performance of LIBs. It has been reported that carbonyl groups on the surface of carbon nanotubes can store lithium ions,17 and materials with a ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CO bond (carbonyl and carboxyl groups) for use in Li-ion batteries as cathode materials have also been proposed.18,19 However, some different opinions have also appeared. Kuo et al.20 thought that the additional capacity is predominantly attributed to phenol groups, but that the carboxyl, lactone and carbonyl groups result in irreversible lithiation/de-lithiation processes. But Wang et al.21 thought that the epoxide group of GO can have a high capacity of 360.4 mA h g−1 at 50 mA g−1 as a cathode material. Therefore, it is a meaningful study to explore the roles of different functional groups of GO in the electrochemical performance of LIBs.
CO bond (carbonyl and carboxyl groups) for use in Li-ion batteries as cathode materials have also been proposed.18,19 However, some different opinions have also appeared. Kuo et al.20 thought that the additional capacity is predominantly attributed to phenol groups, but that the carboxyl, lactone and carbonyl groups result in irreversible lithiation/de-lithiation processes. But Wang et al.21 thought that the epoxide group of GO can have a high capacity of 360.4 mA h g−1 at 50 mA g−1 as a cathode material. Therefore, it is a meaningful study to explore the roles of different functional groups of GO in the electrochemical performance of LIBs.
Thus, in this work, GO with different ratios of functional groups was prepared via low temperature directional thermal reduction technology in a N2 atmosphere. At the same time, nondestructive covalent carboxyl-rich graphene oxide was prepared for the first time by means of adjusting the ultrasonic time, ultrasonic power and the amount of nitric acid. Reduced GO (RGO) with fewer oxygen containing functional groups was also prepared using hydrazine as mentioned in the literature22 to verify the role of the CC double bond.
The prepared GO samples with different ratios of functional groups were investigated and applied as GO based anode materials for LIBs. It is worth noting that the amount of functional groups in the GO is related to the processing temperature. The enhanced reversible capacity of the GO electrode was attributed to the oxygen-containing functional groups and the improved capacities were attributed predominantly to the carbonyl and carboxyl groups, which has been proved by us.
2. Experimental
2.1 Preparation GO and GO electrodes with different ratios of functional groups
All chemical reagents used to prepare GO were analytical grade (purchased from Kelong Chemical Reagent Crop. Chengdu, China) and used as received. GO was synthesized from high-purity natural flake graphite (about 200 meshes, Changsha Shenghua Research Institute, 99.999%) using a modified Hummers method.23,24 A colloidal dispersion of GO in deionized water was prepared with the aid of an ultrasound treatment (20 kHz ultrasound probe) for about 30 min to give a stable amber dispersion. Subsequently, the products were washed with deionized water to remove the extra sulfuric acid and dried under vacuum at 60 °C. The final obtained samples were GO powders. 0.14 g of GO powder was added to 300.0 mL of deionized water and subjected to ultrasound (20 kHz ultrasound probe) treatment for about 30 min to give a stable brown dispersion in a 500 mL beaker. 0.06 g of Super-P (conductive additive, 40 nm, 62 m2 g−1, TIMCAL Graphite & Carbon) was added to the brown dispersion with stirring and sonication for 30 min, then the beaker was placed in an 80 °C water bath to remove the excess water, and at last, a uniform slurry was obtained. The obtained slurry was coated onto Cu foil and dried at 105 °C for 12 h in a vacuum drying oven to remove the adsorbed water on the GO surface, then the foil was cut into disks (10 mm in diameter). The decomposition temperatures of different functional groups are related to the temperature, therefore, GO powders treated with different temperatures (130 °C, 160 °C, 180 °C, 210 °C and 250 °C) in N2 were prepared and investigated as anode materials for LIBs, and the prepared electrodes were named GO-105, GO-130, GO-160, GO-180, GO-210 and GO-250, respectively. At the same time, RGO and carboxyl-rich GO were prepared to demonstrate the roles of the CC bond (CC) and carboxyl (O–CO) group. The RGO was fabricated using hydrazine15,16 and the carboxyl-rich GO was prepared by means of adjusting the ultrasonic time, ultrasonic power and the amount of nitric acid. As is well known, both the thickness and density of working electrodes can obviously affect the electrochemical performance of batteries. To have an accurate comparison, the same thickness and density of working electrodes were prepared.
2.2 Materials characterization
Fourier transform infrared spectroscopy (FT-IR) was used to identify the functional groups of GO treated with different temperatures (Bio-Rad FTS-60VM FT-IR spectrometer). The samples were mixed with KBr and then finely ground to produce a pellet for the FT-IR experiment. X-ray diffraction (XRD) patterns were obtained from an X’Pert MPD DY1219 using Cu/Kα radiation (λ = 1.5406 Å). Thermogravimetric analysis (TGA) was carried out on a TG209F1 (NET-ZSCH, Germany), at a heating rate of 10 °C min−1 from 30 to 600 °C in a nitrogen atmosphere. Raman spectra were obtained using a Jobin-Yvon LabRAM HR 800 UV spectrometer with a 633 nm line of a He–Ne laser as the excitation source. Using Al/Kα radiation (hv = 1486.6 eV), X-ray photoelectron spectroscopy (XPS, PHI5600 Physical Electronics) was used to determine the elemental compositions and assignments of carbon peaks.2.3 Electrochemical measurements
Coin cells were assembled with lithium metal as the counter electrode and a Celgard 2400 was employed as a separator in a glove box filled with Ar gas. The electrolyte was obtained from Capchem. Technology (Shenzhen) Co., Ltd., and consisted of a solution of 1.0 M LiPF6 in ethylene carbonate, dimethyl carbonate, and diethyl carbonate (EC/DMC/DEC, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, in volume). Galvanostatic discharge/charge experiments were carried out on an automatic galvanostatic charge–discharge unit (Land CT 2001A, Wuhan, China) under different current densities from 3.0 to 0.01 V at 25 °C. Electrochemical impedance spectroscopy (EIS) tests were conducted on a PARSTAT 2273 Electrochemical System (Princeton Applied Research, USA). The EIS measurements were performed at an input signal amplitude of 5 mV (vs. open circuit potential) and a frequency ranging from 105 Hz to 10−2 Hz, and the measured data were fitted using Z-View software (Scribner Associates Inc.). All the measurements mentioned above are based on the total mass of the active material (GO).
:1:1, in volume). Galvanostatic discharge/charge experiments were carried out on an automatic galvanostatic charge–discharge unit (Land CT 2001A, Wuhan, China) under different current densities from 3.0 to 0.01 V at 25 °C. Electrochemical impedance spectroscopy (EIS) tests were conducted on a PARSTAT 2273 Electrochemical System (Princeton Applied Research, USA). The EIS measurements were performed at an input signal amplitude of 5 mV (vs. open circuit potential) and a frequency ranging from 105 Hz to 10−2 Hz, and the measured data were fitted using Z-View software (Scribner Associates Inc.). All the measurements mentioned above are based on the total mass of the active material (GO).
3. Results and discussion
Fig. 1a shows the reversible charge/discharge curves of the GO electrodes treated with different temperatures. The charge capacity of GO-105 is 701 mA h g−1 at 0.5C (1C = 372 mA g−1), which is twice that of graphite, and also higher than graphene (700 mA h g−1 in theory and 600 mA h g−1 in reality). However, the discharge capacity of it is over 1600 mA h g−1, so the initial coulomb efficiency of the prepared electrode is only 50%, hindering its large-scale application. Additionally, in the region of 2.5–2.2 V, the GO-105 electrode has a higher irreversible capacity owing to the remaining water (crystal water and the water connected by hydrogen bonds), and this water can cause an electrochemical decomposition reaction in this voltage range.25 With the increase in temperature, the irreversible consumption gradually disappears, and when the temperature increases to 160 °C, the irreversible consumption disappears completely in this area. At the same time, it is observed that the reversible charge capacity increases gradually with increasing temperature, and the GO-180 electrode presents the highest reversible capacity. However, upon further increasing the temperature, the capacity of the GO-210 and GO-250 electrodes begins to decline because most of the capacity is contributed from functional groups which thermally decompose over a certain temperature. | ||
| Fig. 1 The (a) charge and discharge curves and (b) AC impedance spectra with the equivalent circuit from the EIS measurements (inset) of GO treated with different temperatures. | ||
EIS tests were used to further verify the electrochemical performance of the prepared electrodes. Fig. 1b shows the EIS of the GO electrodes treated with different temperatures. These AC impedance spectra are fitted using Z-View software19,25 and the equivalent circuit is shown in Fig. 1b (inset) and its parameters in Table 1. Here Rs is the resistance of the electrolyte, Rct is the charge transfer resistance at the particle/electrolyte interface, W is the Warburg impedance representing the Li-ion diffusion process, the constant phase element (CPE) represents the double-layer capacitance, and C is the insertion capacitance at the applied potential. From Table 1, the Rct of GO treated with different temperatures presents a trend that decreases first and then increases, and the GO-180 electrode has the lowest charge transfer resistance. These findings were consistent with the results of the charge and discharge curves (Fig. 1a).
| Electrodes | Rs (Ω) | Rct (Ω) |
|---|---|---|
| GO-105 | 2.1 | 58 |
| GO-130 | 2.0 | 53 |
| GO-160 | 2.0 | 50 |
| GO-180 | 2.1 | 44 |
| GO-210 | 2.1 | 46 |
| GO-250 | 2.0 | 48 |
To probe the fundamental mechanism for the variation of GO treated with different temperatures, FT-IR was carried out to monitor the variation of the functional groups of the GO with the change in temperature (Fig. 2a). For GO-105, six main adsorption bands are identified, centered at 840, 1050, 1225, 1625, 1730 and 3400 cm−1. The bands at 840 and 1225 cm−1 are assigned to epoxy and epoxide groups,26 the C–O valence vibrations are found at 1050 cm−1 and the O–H of water deformations are at 1400 and 3400 cm−1.27 The weak band at 1730 cm−1 shows some CO stretching vibrations of carbonyl or carboxyl groups, which indicates a small amount of carbonyl and carboxyl groups within the GO.28 The intensities of the peaks at 1250 cm−1, 1050 cm−1, and 840 cm−1 for GO decrease with increasing temperature, especially the band intensities at 840 and 1050 cm−1 which almost disappear when the temperature increases to 180 °C, indicating that the epoxy and epoxide groups are unstable and easily decompose. Additionally, the band at 1625 cm−1 disappeared and a band at 1580 cm−1 appeared at 180 °C, which indicates that all the water (crystal water and the water connected by hydrogen bonds) is removed at this temperature and the CC band intensity recovers somewhat. With the loss of the water and epoxy groups, the intensity of the CO band becomes obviously strengthened at 1730 cm−1.
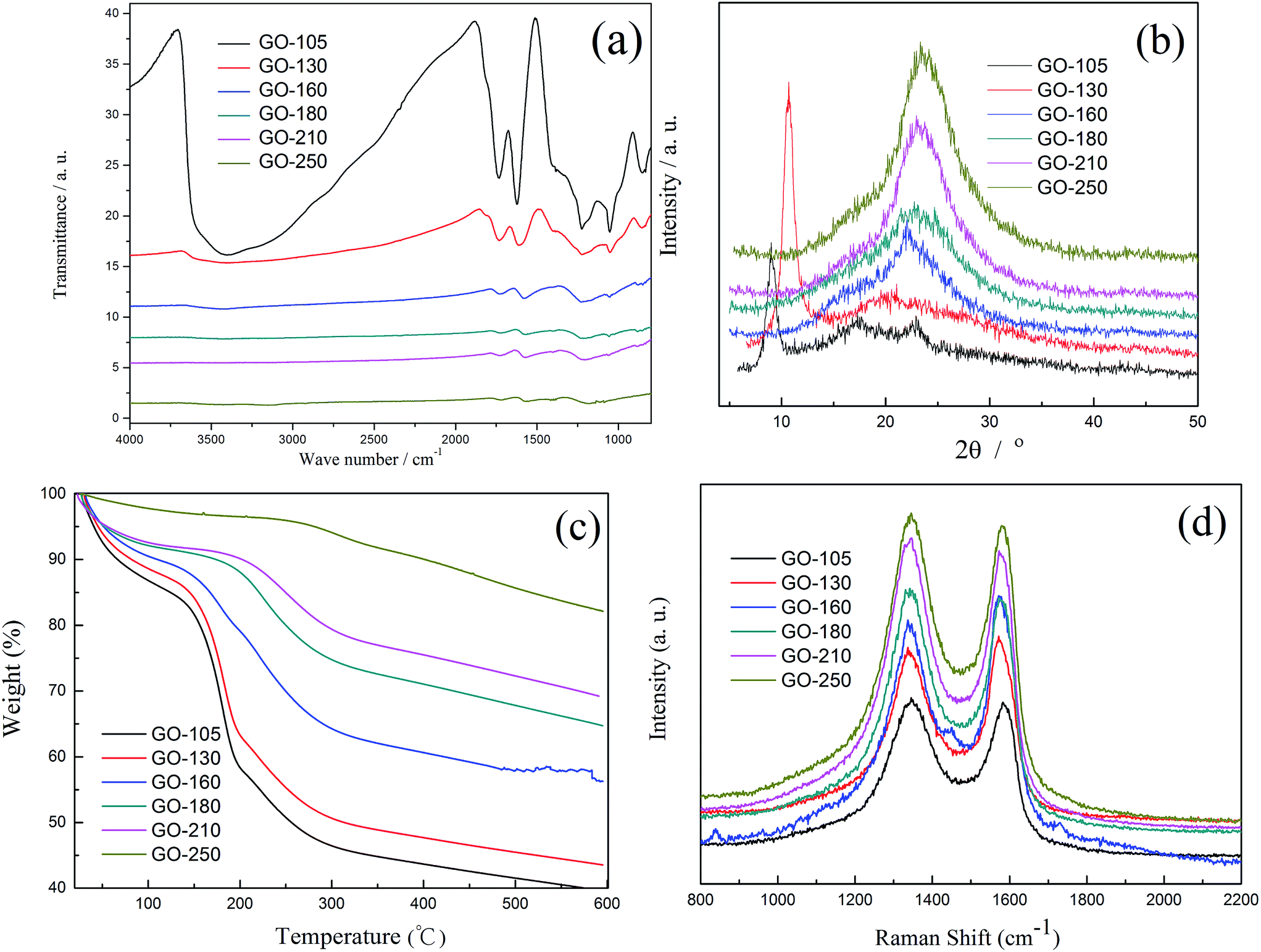 | ||
| Fig. 2 (a) FT-IR, (b) XRD, (c) TGA and (d) Raman spectra of GO treated with different temperatures. | ||
To further verify the conclusions of the FT-IR spectroscopy, XRD was used to characterize the structure of the as prepared electrode materials. Fig. 2b displays the XRD patterns of GO treated with different temperatures. The diffraction peak of exfoliated GO at 2θ = 9.5° (001) features a basal spacing of 0.94 nm (calculated using the Scherrer formula), showing the complete oxidation of graphite to GO due to the introduction of oxygen-containing functional groups onto the graphite sheets.29 With the temperature increasing, there is a slight shift of the diffraction peak towards a higher angle, from 9.5° to 24°, approaching the diffraction peak of graphite at 26.5° (002), and the d-spacing reduces to 0.365 nm, suggesting that the oxygen containing functional groups in-plane of GO, such as hydroxyl and epoxy, have been removed partly.
TGA was used to further assess the change of the structure and functional groups of the GO treated with different temperatures. Fig. 2c displays the TGA plots, which show the weight loss as a function of temperature for different GO (at a heating rate of 5 °C min−1 under a nitrogen atmosphere). The TGA curve of GO-105 exhibits three degradation steps. The first degradation commences at 30 °C, which is attributable to the loss of adsorbed water from the surface of GO. The second degradation step commences at 120 °C due to the loss of hydroxyl and epoxy functional groups and the remaining water molecules.30,31 The third degradation step, starting from 200 °C, involved the pyrolysis of the stabilized oxygen-containing groups. With increasing temperature, significant differences occurred in the second and third degradation steps. When the temperature increased to 180 °C, the first and second degradation steps nearly disappeared, which suggests that the residual water and most of the labile oxygen functionalities (such as the epoxy group) have been removed from the GO at 180 °C. These findings are consistent with the results of the FT-IR tests and XRD patterns. When the temperature increases to 210 °C, the third degradation step commenced, which illustrates that the carboxyl group has begun to decompose and these conclusions are consistent with the results of the charge and discharge curves (Fig. 1a) and the FT-IR tests (Fig. 2a).
The structural integrity of the graphitic materials was confirmed using Raman spectroscopy. As shown in Fig. 2d, GO-105 exhibits a strong D band at ∼1350 cm−1 and a G band at ∼1600 cm−1, corresponding to sp3 and sp2 carbon atoms, respectively. The Raman spectra of GO treated with different temperatures are shown in Fig. 2d. It is worth noting that the D/G intensity ratio (ID/IG) presents a trend that decreases first and then increases (ID/IG = 0.98, 0.97, 0.95, 0.99, and 1.01) because of the loss of adsorbed water and the labile functional groups which can be removed under a low temperature. However, when the temperature increases to 210 °C, new GO domains are created which are smaller in size to the ones present in the initial GO, but are more numerous in number.32
Fig. 3 illustrates the XPS C1s spectra of the GO treated with different temperatures and their curve fittings. Listed in Table 2 are the XPS data in detail, and it can be observed that the content of functional groups varies along with the temperature. The hydroxyl and epoxy groups decrease gradually and even disappear with increasing temperature, and do not conform to the trend of the capacity change with temperature (Fig. 1a). Thus, the conclusion that the hydroxyl and epoxy groups have no contribution to the capacity of GO electrodes is reached. Only the variation of the content of the carboxyl group is consistent with the variation of the specific capacity, as shown in Fig. 1a, testifying that the carboxyl group plays an important role in the specific capacity. However, the roles which the functional groups of the CC bond and carbonyl group play in the electrochemical performance have not been confirmed in detail.
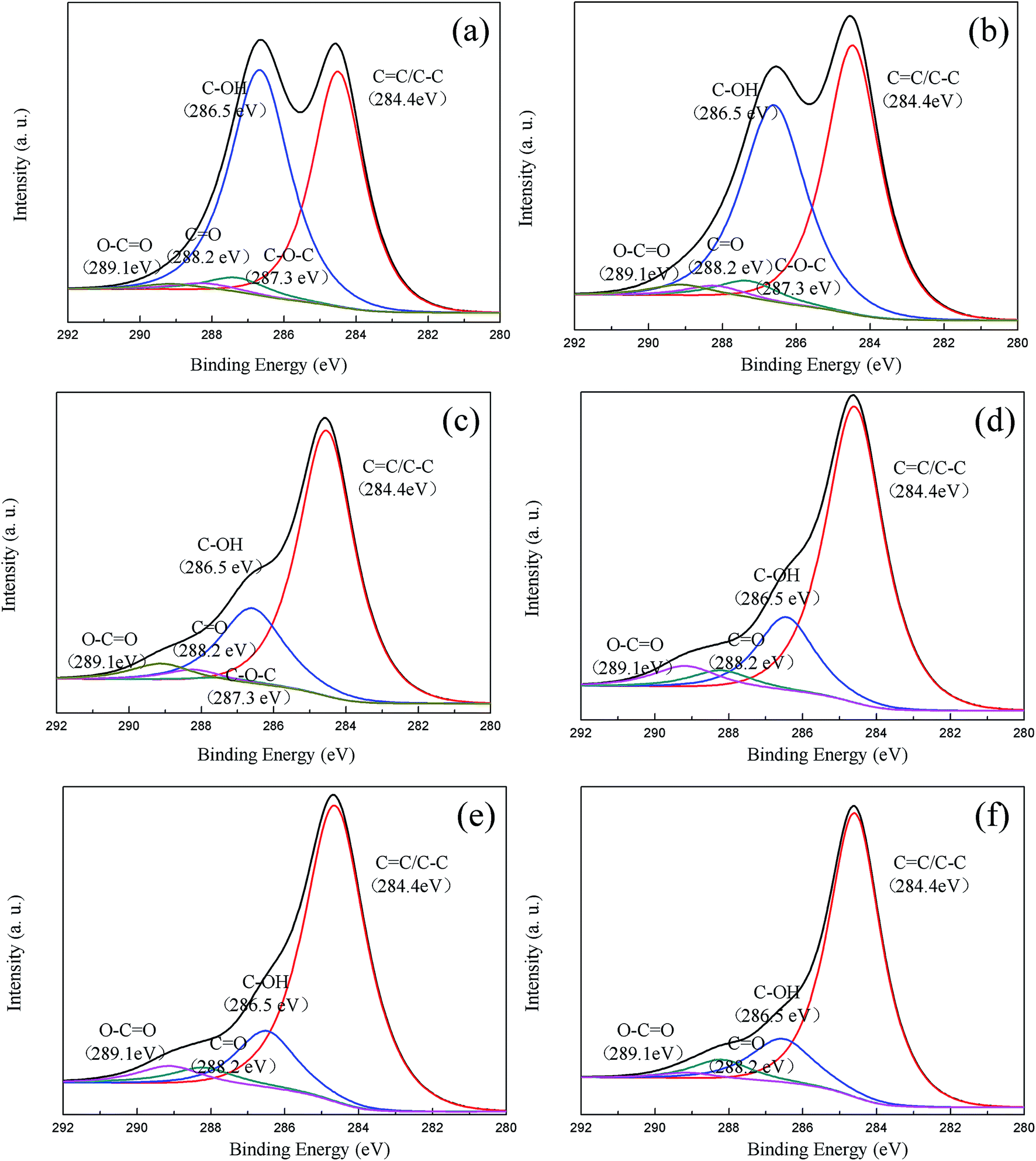 | ||
| Fig. 3 Fitting results of the C1s spectra of the GO treated with different temperatures. | ||
| Samples | C/O ratio | Fitting of C1s (relative atomic percentage, %) | ||||
|---|---|---|---|---|---|---|
| CC/C–C |
C–OH | C–O–C | CO |
O–CO |
||
| GO-105 | 2.03 | 44.9 | 49.1 | 3.4 | 1.5 | 1.5 |
| GO-130 | 2.15 | 48.2 | 43.8 | 3.8 | 1.9 | 2.3 |
| GO-160 | 2.76 | 68.7 | 22.4 | 1.4 | 2.9 | 4.6 |
| GO-180 | 3.44 | 72.0 | 17.8 | 0 | 4.4 | 5.80 |
| GO-210 | 4.06 | 75.7 | 14.4 | 0 | 5.1 | 4.8 |
| GO-250 | 4.88 | 78.2 | 14.3 | 0 | 5.8 | 1.7 |
In order to explore the effect of CC bonds on the electrochemical properties of GO for LIBs, RGO was fabricated using hydrazine. As shown in Fig. 4a, only a small amount of hydroxyl groups is observed and the CC conjugated structure is almost recovered. The rate capability of RGO is better than GO as the CC bond has a better conductive ability. However, the GO has a higher specific capacity than RGO because the GO has more oxygen containing functional groups, which can store more Li ions.16 In conclusion, the CC bond plays an important role in rate performance, but has no contribution to the capacity of GO electrodes for LIBs.
 | ||
| Fig. 4 (a) Fitting results of the C1s spectra of RGO and (b) the rate performance of the GO and RGO electrodes. | ||
XPS was used to detect the variation of functional groups during the charge and discharge processes. The GO-105 electrode before a charge and discharge cycle, and in the lithiated and de-lithiated states was characterized and is shown in Fig. 5. The GO-105 electrode without a charge and discharge cycle test shows five peaks at 284.6, 286.6, 287.3, 288.2 and 289.1 eV, which correspond to sp2 hybridized carbon, hydroxyl (C–OH), epoxide (C–O–C), carbonyl (CO) and carboxyl (O–CO) functional groups, respectively (Fig. 5a). However, certain changes occur in the functional groups after it is subjected to a discharge–charge (lithiation and de-lithiation) process at 0.5C. A comparison of the peaks in Fig. 5a suggests the following facts. First, a new peak of a nearly semi-ionic C–F group arises because of the side reaction of the electrode and electrolyte.18 Second, comparison of the GO-105 electrode before electrochemical testing shows that the peak intensities of the carbonyl (CO) and carboxyl (O–CO) groups have reduced while the C–OH is strengthened obviously after the lithiation process (Fig. 5b). However, there is an inverse process after the delithiation process (Fig. 5c), which proves that the CO (carbonyl and carboxyl) groups and Li+ can undergo a reversible reaction, and these results agree well with the proposed hypothesis that the CO group can improve the reversible charge capacity and initial coulomb efficiency.17–19
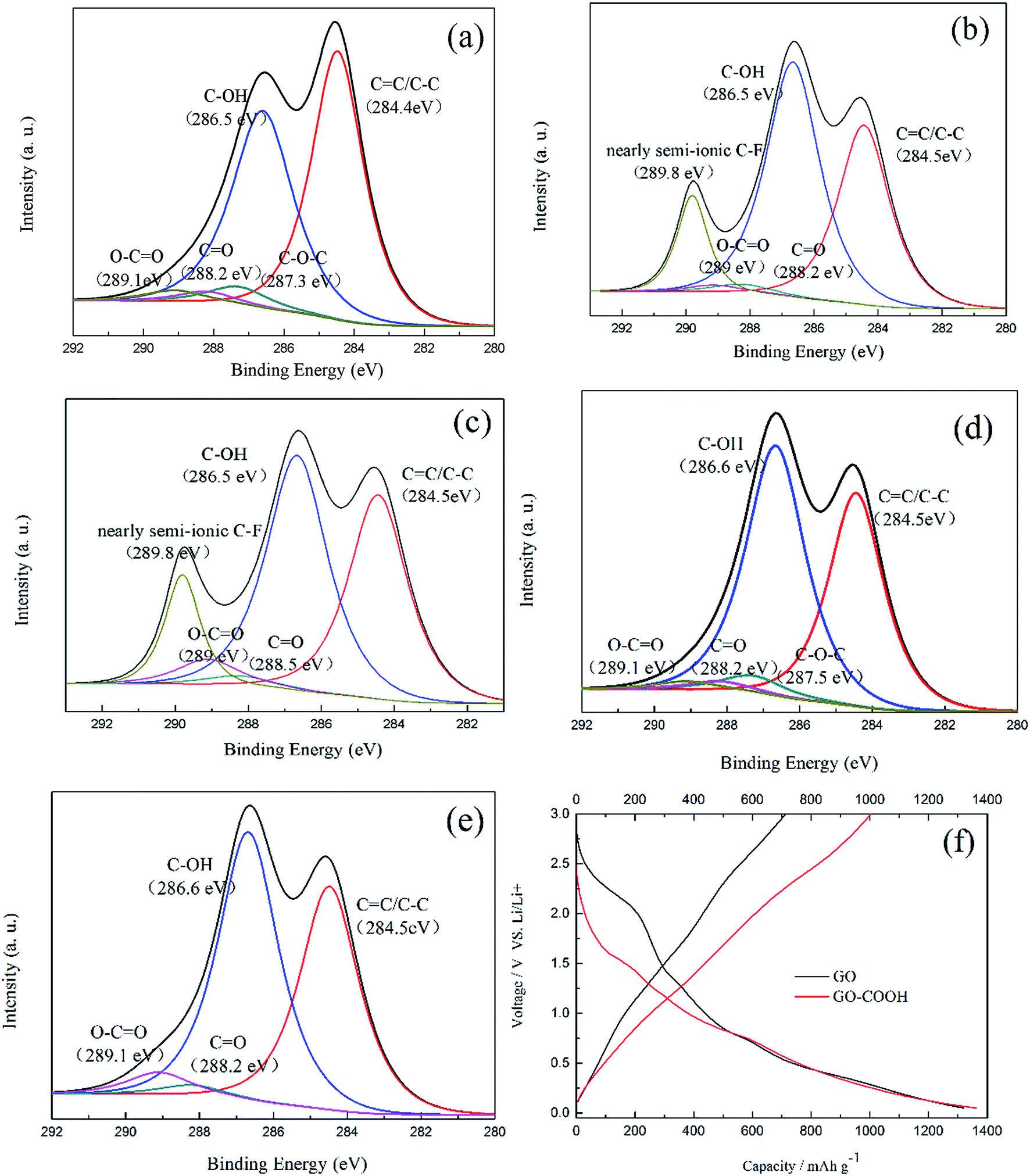 | ||
| Fig. 5 Fitting results of the C1s spectra of the GO-105 electrode (a) before a charge and discharge cycle, (b) in lithiated and (c) in delithiated states, (d) the initial GO and (e) GO–COOH; and (f) the charge and discharge curves of the GO and GO–COOH electrodes. | ||
To prove the hypothesis that the CO group can improve the reversible charge capacity, carboxyl-rich GO was also prepared by means of adjusting the ultrasonic time, ultrasonic power and the amount of nitric acid, and named GO–COOH. Compared with the initial GO, the GO–COOH (Fig. 5e) has a higher content of carbonyl and carboxyl groups, as shown in Table 3. Fig. 5f is the discharge/charge curves of GO and GO–COOH at 0.5C; the GO–COOH electrode, with a higher content of carbonyl and carboxyl groups, has a charge capacity of 1001 mA h g−1, far higher than that of the GO electrode (701 mA h g−1).
| Samples | C/O ratio | Fitting of C1s (relative atomic percentage, %) | ||||
|---|---|---|---|---|---|---|
| CC/C–C |
C–OH | C–O–C | CO |
O–CO |
||
| GO | 2.20 | 40.5 | 52.2 | 3.8 | 1.9 | 1.6 |
| GO–COOH | 2.10 | 40.5 | 52.1 | 0 | 2.1 | 5.3 |
Additionally, the prepared GO–COOH was adopted in (LGC) composite anode materials, which were reported in our previous work.16 The specific capacity of the LTO/GO–COOH (LGCC) composite electrode increased by 10% compared to that of the LGC electrode (shown in Fig. S1a†), and the coulomb efficiency has also been improved greatly from 70% to 82% (shown in Fig. S1b†).
Acknowledgements
This work could not have been accomplished without the financial support from MOST of China (2011CB932604), the National Natural Science Foundation of China (grant no. 51302232) and the 973 Program (grant no. 2013CB934700). Meanwhile our gratitude goes to Analytical and Testing Center of Chengdu Branch, Chinese Academy of Sciences.References
- O. M. Pan, H. B. Wang and Y. H. Jiang, J. Mater. Chem., 2007, 17, 329 RSC.
- S. W. Lee, N. Yabuuchi, B. M. Gallant, S. Chen, B. S. Kim, P. T. Hammond and Y. Shao-Horn, Nat. Nanotechnol., 2010, 5, 531 CrossRef CAS PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359 CrossRef CAS PubMed.
- D. Wang, R. Kou, D. Choi, Z. Yang, Z. Nie, J. Li, L. V. Saraf, D. Hu, J. Zhang, G. L. Graff, J. Liu, M. A. Pope and I. A. Aksay, ACS Nano, 2010, 4, 1587 CrossRef CAS PubMed.
- H. J. Kim, Z. H. Wen, K. H. Yu, O. Mao and J. H. Chen, J. Mater. Chem., 2012, 22, 15514 RSC.
- C. C. Ma, X. H. Shao and D. P. Cao, J. Mater. Chem., 2012, 22, 8911 RSC.
- E. Pollak, B. Geng, K. J. Jeon, I. T. Lucas, T. J. Richardson, F. Wang and R. Kostecki, Nano Lett., 2010, 10, 3386 CrossRef CAS PubMed.
- X. Zhao, C. M. Hayner, M. C. Kung and H. H. Kung, Adv. Energy Mater., 2011, 1, 1079 CrossRef CAS PubMed.
- P. Guo, H. H. Song and X. H. Chen, Electrochem. Commun., 2009, 11, 1320 CrossRef CAS PubMed.
- J. Li, H. Q. Xie and Y. Li, J. Power Sources, 2013, 241, 388 CrossRef CAS PubMed.
- J. Li, H. Q. Xie and Y. Li, J. Nanosci. Nanotechnol., 2015, 15, 3280 CrossRef CAS PubMed.
- B. Xu, S. F. Yue, Z. Y. Sui, X. T. Zhang, S. S. Hou, G. P. Cao and Y. S. Yang, Energy Environ. Sci., 2011, 4, 2826 CAS.
- L. S. Wang, A. N. Lai, C. X. Lin, Q. G. Zhang, A. M. Zhu and Q. L. Liu, J. Membr. Sci., 2015, 492, 58 CrossRef PubMed.
- J. X. Zhang, H. Q. Cao, X. L. Tang, W. F. Fan, G. C. Peng and M. Z. Qu, J. Power Sources, 2013, 241, 619 CrossRef CAS PubMed.
- J. X. Zhang, Z. W. Xie, W. Li, S. Q. Dong and M. Z. Qu, Carbon, 2014, 74, 153 CrossRef CAS PubMed.
- Z. W. Xie, X. Li, W. Li, M. Z. Chen and M. Z. Qu, J. Power Sources, 2015, 273, 754 CrossRef CAS PubMed.
- L. Y. Wang, L. H. Zhuo, H. Y. Cheng, C. Zhang and F. Y. Zhao, J. Power Sources, 2015, 283, 289 CrossRef CAS PubMed.
- B. Z. Jang, C. G. Liu, D. Neff, Z. N. Yu, M. C. Wang, W. Xiong and A. Zhamu, Nano Lett., 2011, 11, 3785 CrossRef CAS PubMed.
- W. Ai, Z. Z. Du, Z. X. Fan, J. Jiang, Y. L. Wang, H. Zhang, L. H. Xie, W. Huang and T. Yu, Carbon, 2014, 76, 148 CrossRef CAS PubMed.
- S. L. Kuo, W. R. Liu, C. P. Kuo, N. L. Wu and H. C. Wu, J. Power Sources, 2013, 244, 552 CrossRef CAS PubMed.
- D. W. Wang, C. H. Sun, G. M. Zhou, F. Li, L. Wen, B. C. Donose, G. Q. Lu, H. M. Cheng and I. R. Gentle, J. Mater. Chem. A, 2013, 1, 3607 CAS.
- L. Yan, R. Y. Li, Z. J. Li, J. K. Liu, Y. J. Fang, G. L. Wang and Z. G. Gu, Electrochim. Acta, 2013, 95, 146 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- N. I. Kovtyukhova, P. J. Ollivier, B. R. Martin, T. E. Mallouk, S. A. Chizhik, E. V. Buzaneva and A. D. Gorchinskiy, Chem. Mater., 1999, 11, 771 CrossRef CAS.
- Z. W. Xie, P. He, L. C. Du, F. Q. Dong, K. Dai and T. H. Zhang, Electrochim. Acta, 2013, 88, 390 CrossRef CAS PubMed.
- C. Hontoria-Lucas, A. J. Lpez-Peinado, J. d. D. Lopez-Gonzlez, M. L. Rojas-Cervantes and R. M. Martn-Aranda, Carbon, 1995, 33, 1585 CrossRef CAS.
- M. M. Hantel, T. Kaspar, R. Nesper, A. Wokaun and R. Kotz, Chem.–Eur. J., 2012, 18, 9125 CrossRef CAS PubMed.
- M. Seredych, J. A. Rossin and T. J. Bandosz, Carbon, 2011, 49, 4392 CrossRef CAS PubMed.
- S. Moussa, G. Atkinson, M. SamyEl-Shall, A. Shehata, K. M. AbouZeid and M. B. Mohamed, J. Mater. Chem., 2011, 21, 9608 RSC.
- T. Kuila, S. Bose, P. Khanra, A. K. Mishra, N. H. Kim and J. H. Lee, Carbon, 2012, 50, 914 CrossRef CAS PubMed.
- Z. J. Fan, W. Kai, J. Yan, T. Wei, L. J. Zhi, J. Feng, Y. M. Ren, L. P. Song and F. Wei, ACS Nano, 2011, 5, 191 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra17854a |
| This journal is © The Royal Society of Chemistry 2015 |