DOI:
10.1039/C5RA17755C
(Paper)
RSC Adv., 2015,
5, 94551-94561
A new heterocyclic skeleton with highly tunable absorption/emission wavelength via H-bonding†
Received
1st September 2015
, Accepted 25th October 2015
First published on 27th October 2015
Abstract
A new heterocyclic system, pyrido[2,1-a]pyrrolo[3,2-c]isoquinoline, was synthesized via Pd-catalyzed intramolecular cyclization of 1-[1-benzyl-2-(2-bromophenyl)-1H-pyrrol-3-yl]pyridin-1-ium bromides. The heterocycles obtained display stimuli responsive fluorescence in solution depending on the nature of the solvent. The strongest blue shift of the emission maxima and growth in luminescence intensity was observed in protic solvents and upon addition of proton donors to solutions of compounds in aprotic solvents. The effect of proton donors on emission characteristics was explained by DFT calculations in terms of H-complex formation with the nucleophilic centres of the molecular skeleton.
Introduction
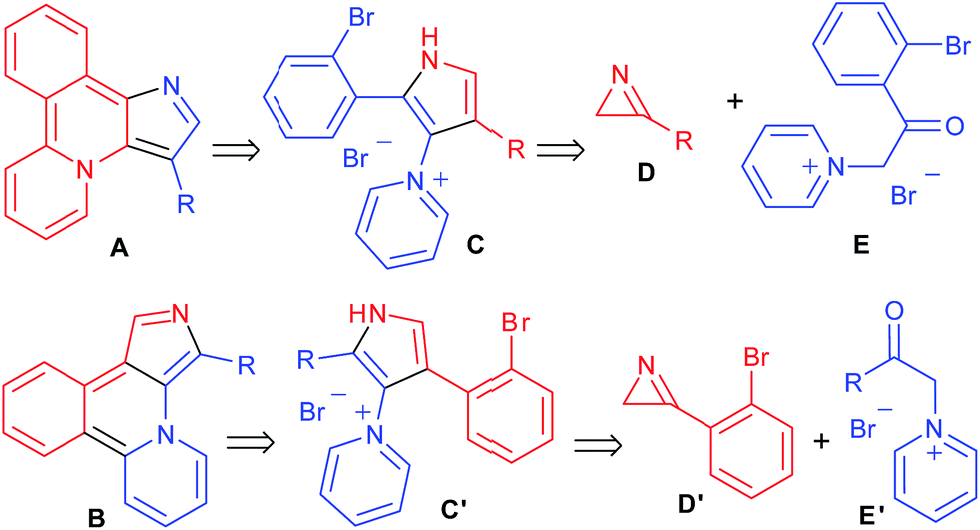
Fluorescence has extremely wide use in various fields of science and technology. In particular, there has been an explosive growth in the application of fluorescence in the biological sciences during recent decades.1 Design of new fluorescent molecular frameworks is, therefore, the subject of ever-growing interest, and often heteroaromatic molecules are the skeletons of choice in the search for novel luminophores. Among them, molecules containing a pyrrole core have a significant importance in the development of the prospective structures.2 Our laboratory has a long standing interest in the synthesis of functionalized pyrrole derivatives.3 Recently a new synthetic route to pyrrolylpyridines was developed by the reaction of pyridinium ylides with 2H-azirines.3h We predicted that o-bromophenyl-substituted pyrrolylpyridines can be used as building blocks for the design of novel heterocyclic systems A (pyrido[2,1-a]pyrrolo[3,2-c]isoquinoline) and B (pyrido[2,1-a]pyrrolo[3,4-c]isoquinoline) according to Scheme 1. The key step of this reaction sequence is the intramolecular cyclization of compounds C/C′, which can be prepared by reaction of the corresponding 2H-azirines D/D′ and N-phenacylpyridinium ylides E/E′. According to calculations at the DFT B3LYP/6-31+G(d,p) level with PCM model for toluene, compound A (R = H) should have long-wave absorption and emission bands at 473 and 642 nm, respectively, whereas compound B (R = H) should have bands at 585 and 829 nm, respectively. Taking into account the particular importance of long wavelength emitters for bioimaging, as well as a possibility to detect strong solvent effects due to the presence of Lewis base centres we decided to synthesize seemingly readily available compounds of the A and B type for spectroscopic investigations.
 |
| | Scheme 1 Retrosynthetic analysis of heterocycles A and B. | |
Results and discussion
The starting 1H-pyrrol-3-yl-pyridin-1-ium bromides 3a,b containing an o-bromophenyl substituent in the correct position for further cyclization to compounds A (R = Ph) and B (R = Ph) under radical or metal-catalyzed conditions were obtained in good yields, using an earlier developed procedure (Table 1, entries 1–2).3h However, attempts to obtain A and B from 3a,b either by radical reactions (Bu3SnH, AIBN, toluene or mixture acetonitrile/toluene, heating up to 90 °C, normal or slow addition of reagents)4 or by palladium-catalyzed reactions5 failed. We therefore converted bromides 3a,b into other possible precursors for the cyclizations, ylides 4a,b, which differ from the salts by the electronic structure, and salts 5a,b, 6 and 7a with the protected pyrrole nitrogen (Table 1, entries 3–8). The attempts to cyclize compounds 4a,b, 5b and 6 were unsuccessful (for more details see ESI†), whereas reactions of 5a and 7a led to certain results.
Table 1 Synthesis of the starting compounds for the cyclizations
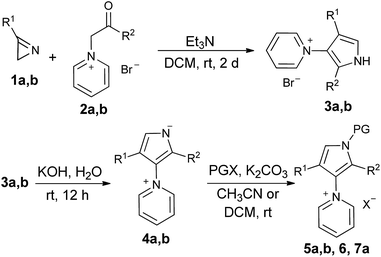
|
| Entry |
R1 |
R2 |
PG |
X |
Yield, % |
| 1 |
Ph |
2-BrC6H4 |
— |
— |
73 (3a) |
| 2 |
2-BrC6H4 |
Ph |
— |
— |
81 (3b) |
| 3 |
Ph |
2-BrC6H4 |
— |
— |
99 (4a) |
| 4 |
2-BrC6H4 |
Ph |
— |
— |
88 (4b) |
| 5 |
Ph |
2-BrC6H4 |
Me |
I |
99 (5a) |
| 6 |
2-BrC6H4 |
Ph |
Me |
I |
72 (5b) |
| 7 |
Ph |
2-BrC6H4 |
Ac |
Cl |
99 (6) |
| 8 |
Ph |
2-BrC6H4 |
Bn |
Br |
97 (7a) |
In the case of pyridinium iodide 5a, traces of the expected product were detected under radical conditions and finally product 8 (for X-ray see ESI†) was isolated in low yield when the cyclization was carried out in the presence of Pd(OAc)2. The benzylated substrate 7a cyclized to the desired product 9a in a good yield under the same conditions (Scheme 2).
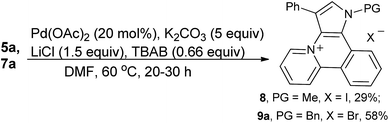 |
| | Scheme 2 Cyclization of compounds 5a and 7a. | |
Debenzylation of compound 9a was attempted using two different conditions: with hydrogen on Pd/C or with Adam's catalyst. Both reactions led, however, to a partial reduction of the aromatic system 9a to give 10, according to NMR and mass-spectra, but left the benzyl group intact (Scheme 3). To remove the benzyl group and at the same time to avoid the reduction of the heterocyclic core in 9a the action of AlCl3 in refluxing benzene was tested. To our surprise instead of expected compound A (R = Ph) the product 11 was obtained in quantitative yield. Not only the benzyl group was removed but also the phenyl group on the pyrrole ring migrated from the β- to the α-position (Scheme 3). To the best of our knowledge there are only two examples of similar migration at a pyrrole ring under the action of Lewis acids.6
 |
| | Scheme 3 Deprotection of compound 9a. | |
The structure of 11 was undoubtedly confirmed by X-ray analysis (Fig. 1). To be sure that the migration occurred in the last stage of the synthesis an X-ray analysis of 9a was also performed.
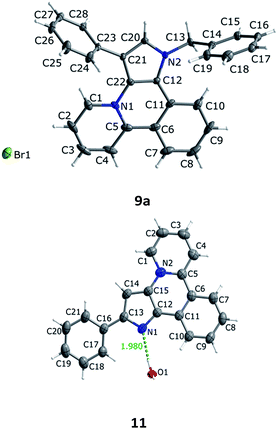 |
| | Fig. 1 Molecular structure of compounds 9a and 11. Carbon, nitrogen and oxygen atoms are grey, blue and red, respectively. Bromine is green. | |
To modify the spectroscopic properties of the pyrido[2,1-a]pyrrolo[3,2-c]isoquinoline system compounds 12 and 13, with additional phenyl groups at the pyrrole or at the pyridine rings, were synthesized using the protocol developed, see Scheme 4. Again, X-ray analysis (Fig. 2) confirmed that deprotection of 9c results in migration of the phenyl group at the pyrrole ring to give the product 13.
 |
| | Scheme 4 Synthesis of compounds 12 and 13. | |
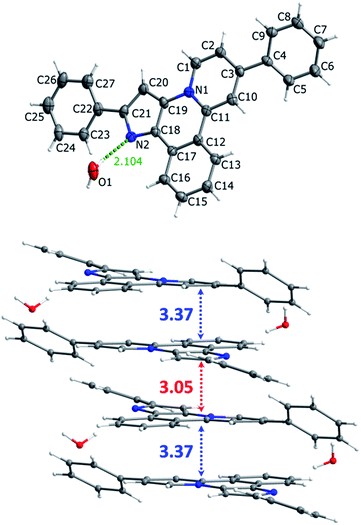 |
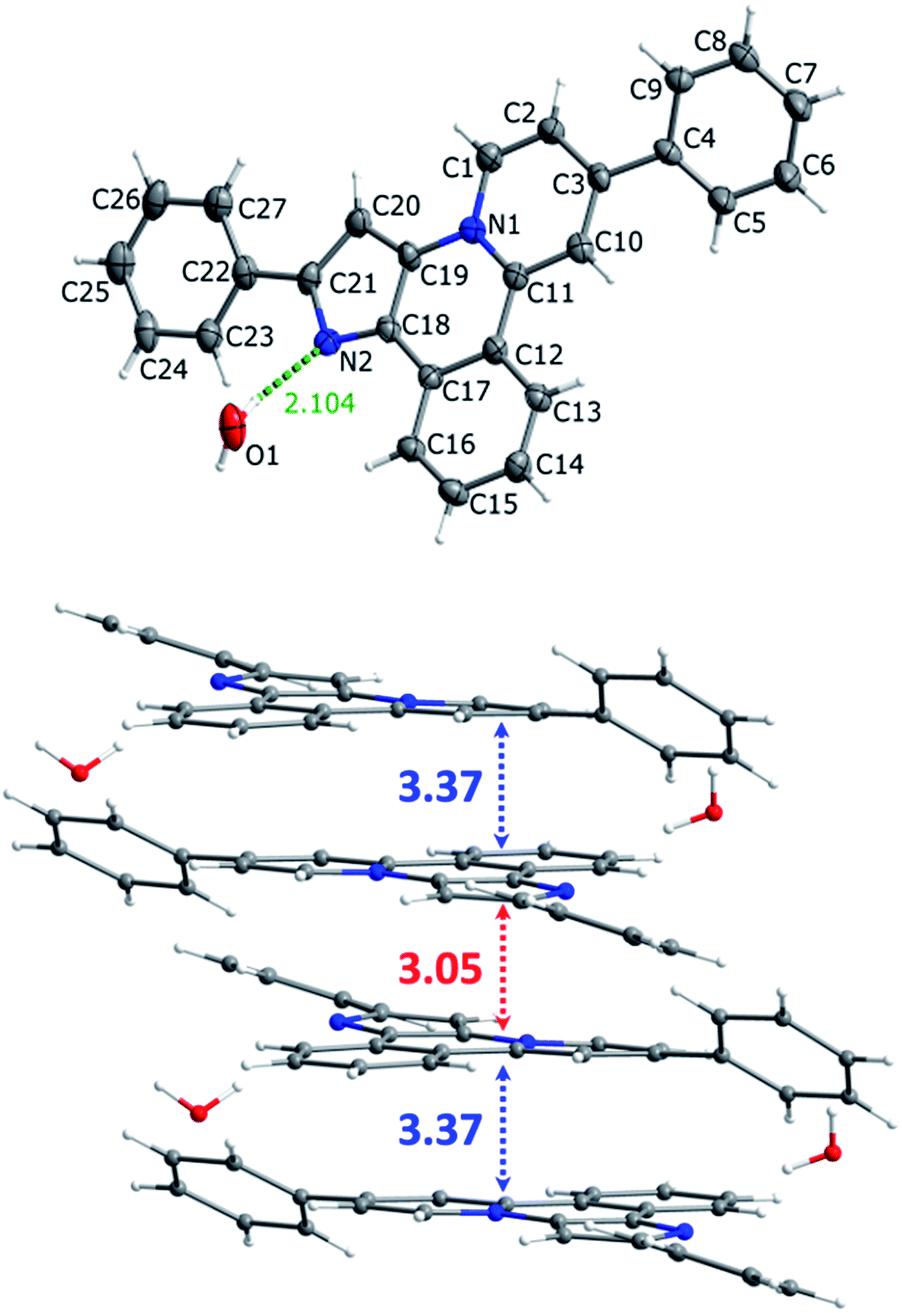
| | Fig. 2 Molecular structure (top) and crystal packing (bottom) of compound 13. Carbon, nitrogen and oxygen atoms are grey, blue and red, respectively. | |
Molecular structure and crystal cell packing of 13 are shown in Fig. 2, crystallographic data and structural parameters of this molecule together with the corresponding data for 8, 9a, 11 are given in (ESI), Table S1–S29.† Major structural parameters of these molecules fall in the range typical for this type of compounds. Essentially planar tetracyclic aromatic backbones make possible their π-stacking in solid state with two substantially different (3.05 and 3.37 Å) distances between the planes. This variation is due to insertion of co-crystallized water molecules into the crystal structure to give bigger separation between two adjacent layers, see Fig. 2. Similar packing motif was observed for the other compounds studied.
The compounds obtained are luminescent in solutions, with the quantum yields amounting up to 81% in the case of 13 in methanol. Photophysical data are given in Table 2 and representative examples of absorption, excitation and emission spectra in Fig. 3, S1–S3 (ESI).† Typically small values of Stokes shifts together with excited state lifetime in nanosecond domain clearly indicate that the emission observed originates from the singlet excited state, i.e. fluorescence. Analogously to the previously studied compounds with similar structures containing a quinolizinium nitrogen7 the emission can be assigned to the singlet–singlet π*–π transitions between the orbitals of fused aromatic system. This was also confirmed by theoretical calculations, vide infra.
Table 2 Photophysical characteristics of 11–13 in toluene, acetonitrile, dichloromethane (DCM) and methanol solutions at room temperature, λex = 420 nm. Lifetimes (τ) were measured at λmax of the emission bands
| |
Solvent |
Absorbance λmax, nm (ε, 10−3 M−1 cm−1) |
Emission λmax, nm |
Excitation λmax, nm |
τ, ns |
QY, % |
| 11 |
MePh |
297(33); 325(26.5)sh; 334(29); 423(8); 512(10) |
602 |
337, 430, 516 |
4.6 ± 0.5 |
14 |
| MeCN |
288(30.5); 320(30.5)sh; 328(32); 404(6); 491(12) |
598 |
405, 494 |
3.9 ± 0.5 |
8 |
| DCM |
293(33); 321(30)sh; 332(34); 413(7); 500(12) |
590 |
329, 413, 502 |
5.5 ± 0.5 |
18 |
| MeOH |
295(35); 326(11)sh; 426(13) |
507 |
295, 428 |
9.0 ± 0.5 |
76 |
| 12 |
MePh |
297(42); 331(45); 417(12); 521(14) |
640 |
427, 528 |
1.0 ± 0.2 |
7 |
| MeCN |
287(48); 323(47); 401(13); 497(15) |
630 |
401, 498 |
1.0 ± 0.2 |
0.7 |
| DCM |
291(46); 327(42); 409(12); 509(14) |
628 |
438, 500sh |
1.0 ± 0.2 |
3 |
| MeOH |
293(59); 426(18.5) |
530 |
292, 426 |
10.3 ± 0.5 |
47 |
| 13 |
MePh |
306(45); 338(42); 429(11.4); 532(19) |
645 |
440, 539 |
1.4 ± 0.2 |
7.5 |
| MeCN |
300(68.5); 335(69.5); 419(19); 514(35) |
642 |
417, 513 |
1.5 ± 0.2 |
2.5 |
| DCM |
301(62); 337(50); 425(14); 522(25) |
626 |
330, 420, 522 |
1.7 ± 0.2 |
9 |
| MeOH |
385(81); 443(38) |
537 |
443 |
7.5 ± 0.5 |
81 |
 |
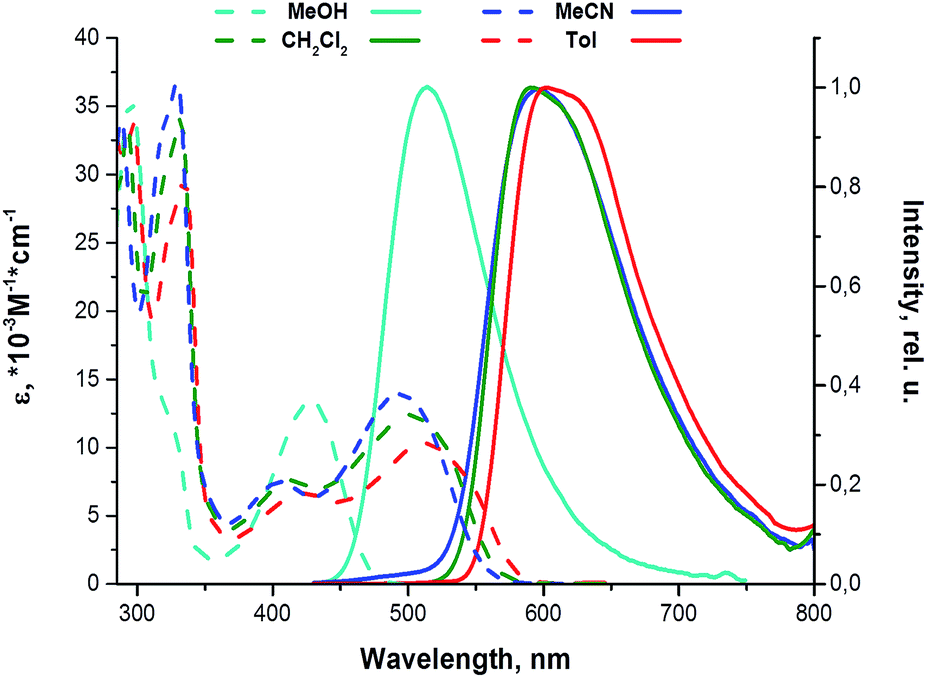
| | Fig. 3 Room temperature absorption and emission spectra of 11 in different solvents. | |
The compounds studied display rather small and nonsystematic variations in absorption and emission characteristics (Δ ca. 10–15 nm) in response to the changes in solvent polarity for aprotic solvents, (ε = 2.9, 8.3, 37.5 for toluene, dichloromethane and acetonitrile, respectively), Fig. 3 and Table 2. However they feature a strong stimuli-responsive behavior in the presence of proton donors to give a considerable blue shift in the absorption and emission spectra of their solution in neat methanol (ε = 32.6), which is a protic solvent and may form a hydrogen bonds with the nucleophilic centers in the aromatic skeleton of the molecules under study.
Titration of aprotic acetonitrile solution of 11 with methanol (Fig. 4 and 5) shows clear cut isosbestic points in the absorption and emission spectra that is indicative of chemical equilibrium between solvated (with methanol) and unsolvated forms of 11–13.
 |
| | Fig. 4 Titration of 11 (4.25 × 10−5 mmol) with methanol, room temperature. | |
 |
| | Fig. 5 Dependence of absorbance (424 nm) and emission (515 nm) intensity of 11 (4.25 × 10−5 mmol, acetonitrile/methanol solution) on the concentration of methanol. | |
A very similar spectroscopic pattern was observed upon titration of an acetonitrile solution of 11 with formic acid and reversed titration of the acidified sample with DBU (1,8-diazabicycloundec-7-ene), Fig. 6 and 7.
 |
| | Fig. 6 Titration of 11 (6.4 × 10−5 mmol) with HCOOH in acetonitrile solution. | |
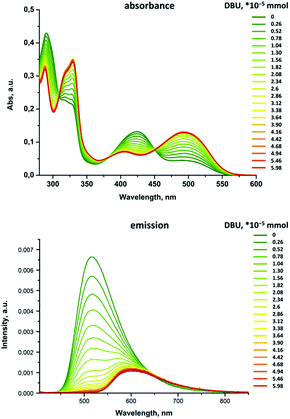 |
| | Fig. 7 Titration of 11·HCl (5.29 × 10−5 mmol) with DBU in acetonitrile solution. | |
It is worth noting that the reaction of 11 with both methanol and formic acid results also in a five-fold increase in the emission intensity that paves the way to applications of these compounds in sensing. The blue shift of the absorption and emission bands can be explained by stabilization of the molecular ground state due to either protonation of the nucleophilic centers or hydrogen bond formation, in particular, at the pyrrole nitrogen. This induces a partial positive charge onto the polyconjugated aromatic chromophore that was also confirmed by the quantum chemical calculations, described below.
The quantum chemical calculations at the TD DFT B3LYP/6-31+G(d,p) PCM level of theory were performed for 11 (Table 3 and ESI†) to establish the reasons for the strong influence of proton donors on the absorbance and emission. The calculation results are in good agreement with experiment for the absorbance and emission in various aprotic solvents (Table 3, entries 1–3), but gave considerable deviation for methanol (Table 3, entry 4). In order to find a fit with the experimental data the H-complexes of 11 with a different (1–5) number of MeOH molecules bound to the negatively charged sites were calculated. It was found that formation of H-complexes leads to the blue shift of the calculated absorption and emission bands up to three MeOH molecules, and then reaches a plateau (Table 3, entries 5–7, Fig. 8, ESI Table S32†). This evidently indicates saturation in the H-complex formation. The calculated values of absorption and emission maxima now fit well with experimental values. The observed blue shift is due to the higher ground state stabilization upon H-complex formation compared to the first exited state (Fig. 8, ESI Table S32†). Good agreement with experiment was also obtained for protonated and solvated forms of 11 in MeCN solution (Table 3, entries 6 and 7).
Table 3 Absorption (exp./calcd (f)) and emission (exp./calcd) of 11, nm
| Entry |
Solvent |
Absorption |
Emission |
| 1 |
Toluene |
512/506 (0.311) |
602/606 |
| 2 |
DCM |
500/489 (0.311) |
— |
| 3 |
MeCN |
491/481 (0.303) |
598/571 |
| 4 |
MeOH |
426/481 (0.299) |
507/572 |
| 5 |
MeOH, complex 3·MeOH |
426/453 (0.315) |
507/538 |
| 6 |
MeCN, complex HCO2H |
424/437 (0.300) |
515/530 |
| 7 |
MeCN, complex 3·MeOH |
424/453 (0.318) |
515/538 |
 |
| | Fig. 8 Frontier MO of 11 and complexes of 11 with MeOH (DFT B3LYP/6-31+G(d,p), PCM for MeOH). | |
Conclusions
A new heterocyclic skeleton, pyrido[2,1-a]pyrrolo[3,2-c]isoquinoline, was synthesized via Pd-catalyzed intramolecular cyclization of 1-[1-benzyl-2-(2-bromophenyl)-1H-pyrrol-3-yl]pyridin-1-ium bromides, while the cyclization under the radical conditions failed. Removal of the benzyl protecting group using AlCl3, was accompanied by the migration of the aryl group from the C-3 to C-2 of the heterocyclic core. Depending on the nature of solvent the compounds obtained display moderate to strong fluorescence in solution giving some variations in emission maxima position upon changes in solvent polarity, but the strongest effect was observed in methanol solutions and upon addition of proton donors to aprotic solvents. In both cases the observed blue shift of emission maxima was accompanied by a substantial growth in luminescence intensity. These observations clearly indicate a prospective for applications of the compounds based on this heterocyclic skeleton in sensing. Theoretical calculations assign the emission to a π–π* transition in the polyaromatic core. The effect of protonation and protic solvents was adequately explained in terms of H-complex formation with the nucleophilic centres of the molecular skeleton.
Experimental
General
Melting points were determined on a capillary melting point apparatus Stuart® SMP30. 1H (400 MHz) and 13C (100 MHz) NMR spectra were determined in CDCl3 or DMSO-d6 with Bruker AVANCE III 400. Chemical shifts (δ) are reported in parts per million downfield from tetramethylsilane (TMS δ = 0.00); 1H NMR spectra were calibrated according to the residual peak of CDCl3 (7.26 ppm) or DMSO-d6 (2.50 ppm). For all new compounds 13C{1H} and 13C DEPT135 were recorded and calibrated according to the peak of CDCl3 (77.00 ppm) or DMSO-d6 (39.51 ppm). Mass spectra were recorded on a Bruker maXis HRMS-ESI-QTOF, electrospray ionization, positive mode. IR-spectra were recorded on a Bruker FT-IR spectrometer Tensor 27 for tablets in KBr, only characteristic absorption is indicated. Thin-layer chromatography (TLC) was conducted on aluminium sheets with 0.2 mm silica gel (fluorescent indicator, Macherey-Nagel).
General procedure for the synthesis of 1-pyrrol-3-yl-pyridin-1-ium bromides 3a–d
To a stirred suspension of 1-(2-aryl-2-oxoethyl)pyridin-1-ium bromide (5 mmol) 2 and 2H-azirine 1 (7.5 mmol, 1.5 equiv.) in dichloromethane (DCM) (15 mL) triethylamine (758 mg, 7.5 mmol, 1.5 equiv.) was added dropwise, and then the reaction mixture was stirred at rt for 2 days. After the reaction was completed the precipitate was collected, washed with DCM (3 × 3 mL) and dried to obtain an analytically pure product.
1-(2-(2-Bromophenyl)-4-phenyl-1H-pyrrol-3-yl)pyridin-1-ium bromide (3a). Bright-yellow solid, mp 281–282 °C, yield 1.55–1.66 g, 68–73%, obtained from 1-(2-(2-bromophenyl)-2-oxoethyl)pyridin-1-ium bromide (2a) (1.785 g, 5 mmol), 3-phenyl-2H-azirine (1a) (878 mg, 7.5 mmol, 1.5 equiv.) and triethylamine (758 mg, 7.5 mmol, 1.5 equiv.). 1H NMR (DMSO-d6): δ 7.08 (d, J = 7.1 Hz, 2H), 7.23–7.28 (m, 1H), 7.28–7.34 (m, 2H), 7.37–7.42 (m, 1H), 7.47–7.53 (m, 1H), 7.50 (s, 1H), 7.58 (dd, J = 7.6 Hz, J = 1.3 Hz, 1H), 7.68 (d, J = 8.0 Hz, 1H), 8.11–8.19 (m, 2H), 8.65–8.73 (m, 1H), 8.91 (d, J = 5.7 Hz, 2H), 12.37 (s, 1H). 13C NMR (DMSO-d6): δ 117.8 (CH), 118.7 (C), 123.2 (C), 123.5 (C), 126.5 (C), 126.9 (CH), 126.9 (CH), 128.2 (CH), 128.6 (CH), 129.1 (CH), 129.4 (C), 131.1 (C), 131.5 (CH), 132.9 (CH), 133.3 (CH), 146.8 (CH), 147.0 (CH). HRMS (ESI) m/z: 375.0491 calcd for C21H16BrN2+ [M − Br]+, found 375.0492. IR (KBr, cm−1): ν 3055, 1623, 1602, 1466, 1445, 752.
1-(4-(2-Bromophenyl)-2-phenyl-1H-pyrrol-3-yl)pyridin-1-ium bromide (3b). Orange solid, mp 351–352 °C, yield 1.109 g, 81%, obtained from 1-(2-oxo-2-phenylethyl)pyridin-1-ium bromide (2b) (834 mg, 3 mmol), 3-(2-bromophenyl)-2H-azirine (1b) (882 mg, 4.5 mmol, 1.5 equiv.) and triethylamine (455 mg, 4.5 mmol, 1.5 equiv.). 1H NMR (DMSO-d6): δ 7.20 (d, J = 6.8 Hz, 2H), 7.24–730 (m, 1H), 7.33 (s, 1H), 7.35–7.46 (m, 5H), 7.60 (d, J = 8.0 Hz, 1H), 8.10–8.18 (m, 2H), 8.65–8.73 (m, 1H), 8.92 (d, J = 5.7 Hz, 2H), 12.46 (s, 1H). 13C NMR (DMSO-d6): δ 118.9 (CH), 119.3 (C), 123.0 (C), 123.7 (C), 126.3 (C), 126.8 (CH), 128.1 (CH), 128.34 (CH), 128.35 (C), 128.6 (CH), 129.3 (CH), 130.0 (CH), 132.0 (C), 132.6 (CH), 133.0 (CH), 147.0 (CH), 147.1 (CH). HRMS (ESI) m/z: 375.0491 calcd for C21H16BrN2+ [M − Br]+, found 375.0510. IR (KBr, cm−1): ν 3405, 3118, 1623, 1472.
1-(2-(2-Bromophenyl)-4,5-diphenyl-1H-pyrrol-3-yl)pyridin-1-ium bromide (3c). Dark-yellow solid, mp 255–258 °C, yield 2.022 g, 76%, obtained from 1-(2-(2-bromophenyl)-2-oxoethyl)pyridin-1-ium bromide (1a) (1.785 g, 5 mmol), 2,3-diphenyl-2H-azirine (1c) (1.448 g, 7.5 mmol, 1.5 equiv.) and triethylamine (758 mg, 7.5 mmol, 1.5 equiv.). 1H NMR (DMSO-d6): δ 7.12–7.17 (m, 2H), 7.26–7.44 (m, 9H), 7.50–7.56 (m, 1H), 7.68 (dd, J = 7.7 Hz, J = 1.5 Hz, 1H), 7.71 (dd, J = 8.1 Hz, J = 0.9 Hz, 1H), 8.06–8.12 (m, 2H), 8.58–8.65 (m, 1H), 8.88 (d, J = 5.9 Hz, 2H), 12.55 (s, 1H). 13C NMR (DMSO-d6): δ 116.8 (C), 123.5 (C), 125.5 (C), 125.9 (C), 127.3 (CH), 127.6 (CH), 127.7 (CH), 128.1 (CH), 128.2 (CH), 128.6 (CH), 128.7 (C), 129.0 (CH), 129.1 (C), 129.9 (CH), 130.8 (C), 131.0 (C), 131.5 (CH), 132.9 (CH), 133.5 (CH), 146.8 (CH), 146.9 (CH). HRMS (ESI) m/z: 451.0804 calcd for C27H20BrN2+ [M − Br]+, found 451.0809. IR (KBr, cm−1): ν 3514, 3440, 3035, 1630, 1601, 1476, 768, 703, 676.
1-(2-(2-Bromophenyl)-4-phenyl-1H-pyrrol-3-yl)-4-phenylpyridin-1-ium bromide (3d). Bright-yellow solid, mp > 300 °C, yield 1.863 g, 70%, obtained from 1-(2-(2-bromophenyl)-2-oxoethyl)-4-phenylpyridin-1-ium bromide (2c) (2.165 g, 5 mmol), 3-phenyl-2H-azirine (1a) (878 mg, 7.5 mmol, 1.5 equiv.) and triethylamine (758 mg, 7.5 mmol, 1.5 equiv.). 1H NMR (DMSO-d6): δ 7.16 (d, J = 7.0 Hz, 2H), 7.23–7.29 (m, 1H), 7.30–7.36 (m, 2H), 7.37–7.43 (m, 1H), 7.48–7.54 (m, 2H), 7.58–7.72 (m, 5H), 8.11 (d, J = 7.0 Hz, 2H), 8.53 (d, J = 7.1 Hz, 2H), 8.89 (d, J = 7.0 Hz, 2H), 12.34 (s, 1H). 13C NMR (DMSO-d6): δ 117.8 (CH), 118.8 (C), 122.7 (C), 123.5 (C), 124.5 (CH), 126.6 (C), 126.9 (CH), 127.0 (CH), 128.2 (CH), 128.4 (CH), 129.1 (CH), 129.6 (C), 129.7 (CH), 131.5 (C), 131.5 (CH), 132.7 (C), 132.8 (CH), 132.9 (CH), 133.3 (CH), 146.6 (CH), 155.2 (C). HRMS (ESI) m/z: 451.0804 calcd for C27H20BrN2+ [M − Br]+, found 451.0816. IR (KBr, cm−1): ν 3496, 3130, 1633, 1436, 1216.
General procedure for the synthesis of 3-(pyridin-1-ium-1-yl)pyrrol-1-ides 4a–d
A suspension of 1-pyrrol-3-yl-pyridin-1-ium bromides 3 (4 mmol) in aq. solution of KOH (448 mg, 8 mmol, 2 equiv., 50 mL H2O) was sonicated for 30 min and then vigorously stirred for 12 h. The precipitate was filtered, washed with water, and thoroughly dried to obtain analytically pure pyrrolide 4 in almost quantitative yields.
2-(2-Bromophenyl)-4-phenyl-3-(pyridin-1-ium-1-yl)pyrrol-1-ide (4a). Bright-orange solid, mp 208 °C, yield 1.486 g, 99%, obtained from 1-(2-(2-bromophenyl)-4-phenyl-1H-pyrrol-3-yl)pyridin-1-ium bromide (3a) (1.824 g, 4 mmol) and KOH (448 mg, 8 mmol, 2 equiv.). 1H NMR (DMSO-d6): δ 6.88–6.98 (m, 3H), 6.98–7.06 (m, 1H), 7.06–7.15 (1H), 7.15–7.24 (m, 2H), 7.31–7.38 (m, 1H), 7.41 (d, J = 7.9 Hz, 1H), 7.57 (d, J = 7.2 Hz, 1H), 7.85–7.98 (m, 2H), 8.30–8.39 (m, 1H), 8.49 (d, J = 5.6 Hz, 2H). 13C NMR (DMSO-d6): δ 117.0 (C), 122.16 (C), 122.2 (C), 123.9 (CH), 125.7 (CH), 127.5 (CH), 127.7 (CH), 127.74 (CH), 128.7 (CH), 129.4 (CH), 132.1 (CH), 133.6 (CH), 134.5 (C), 135.8 (C), 138.2 (C), 142.2 (CH), 145.3 (CH). HRMS (ESI) m/z: 375.0491 calcd for C21H16BrN2+ [M + H]+, found 375.0503. IR (KBr, cm−1): ν 3053, 1596, 1526, 764.
4-(2-Bromophenyl)-2-phenyl-3-(pyridin-1-ium-1-yl)pyrrol-1-ide (4b). Cherry-red solid, mp 166–168 °C, yield 1.321 g, 88%, obtained from 1-(4-(2-bromophenyl)-2-phenyl-1H-pyrrol-3-yl)pyridin-1-ium bromide (3b) (1.824 g, 4 mmol) and KOH (448 mg, 8 mmol, 2 equiv.). 1H NMR (DMSO-d6): δ 6.82 (s, 1H), 6.98–7.09 (m, 2H), 7.11–7.19 (m, 4H), 7.19–7.24 (m, 1H), 7.24–7.31 (m, 1H), 7.49 (dd, J = 8.0 Hz, J = 0.8 Hz, 1H), 7.91–8.0 (m, 2H), 8.38–8.46 (m, 1H), 8.64 (d, J = 5.6 Hz, 2H). 13C NMR (DMSO-d6): δ 118.1 (C), 122.8 (C), 123.3 (C), 124.2 (CH), 125.5 (CH), 127.1 (CH), 127.7 (CH), 127.8 (CH), 128.2 (CH), 129.9 (CH), 132.0 (CH), 132.6 (CH), 132.7 (C), 136.5 (C), 137.1 (C), 142.9 (CH), 146.0 (CH). HRMS (ESI) m/z: 375.0491 calcd for C21H16BrN2+ [M + H]+, found 375.0502. IR (KBr, cm−1): ν 3437, 3106, 3054, 1595, 1523, 1471, 1453.
2-(2-Bromophenyl)-4,5-diphenyl-3-(pyridin-1-ium-1-yl)pyrrol-1-ide (4c). Bright-red solid, mp 232 °C, yield 1.769 g, 98%, obtained from 1-(2-(2-bromophenyl)-4,5-diphenyl-1H-pyrrol-3-yl)pyridin-1-ium bromide (3c) (2.129 g, 4 mmol) and KOH (448 mg, 8 mmol, 2 equiv.). 1H NMR (DMSO-d6): δ 6.92–6.98 (m, 1H), 7.02–7.19 (m, 6H), 7.19–7.26 (m, 2H), 7.35–7.46 (m, 4H), 7.69 (dd, J = 7.7 Hz, J = 1.6 Hz, 1H), 7.80–7.87 (m, 2H), 8.24–8.31 (m, 1H), 8.35 (d, J = 5.7 Hz, 2H). 13C NMR (DMSO-d6): δ 115.7 (C), 121.9 (C), 123.6 (CH), 125.0 (C), 125.5 (CH), 126.4 (CH), 127.35 (CH), 127.4 (CH), 127.5 (CH), 127.8 (CH), 128.6 (CH), 129.9 (CH), 132.2 (CH), 133.4 (C), 133.7 (CH), 136.3 (C), 137.0 (C), 137.8 (C), 139.5 (C), 142.0 (CH), 145.1 (CH). HRMS (ESI) m/z: 451.0804 calcd for C27H20BrN2+ [M + H]+, found 451.0815. IR (KBr, cm−1): ν 3055, 1597, 1496, 1480, 1440, 766.
2-(2-Bromophenyl)-4-phenyl-3-(4-phenylpyridin-1-ium-1-yl)pyrrol-1-ide (4d). Dark-red solid, mp > 300 °C, yield 1.788 g, 99%, obtained from 1-(2-(2-bromophenyl)-4-phenyl-1H-pyrrol-3-yl)-4-phenyl-pyridin-1-ium bromide (3d) (2.128 g, 4 mmol) and KOH (448 mg, 8 mmol, 2 equiv.). 1H NMR (DMSO-d6): δ 7.13 (d, J = 7.1 Hz, 2H), 7.19–7.25 (m, 1H), 7.27–7.37 (m, 3H), 7.39 (s, 1H), 7.45–7.51 (m, 1H), 7.57–7.68 (m, 5H), 8.10 (d, J = 6.9 Hz, 2H), 8.49 (d, J = 7.1 Hz, 2H), 8.81 (d, J = 7.0 Hz, 2H). 13C NMR (DMSO-d6): δ 118.5 (C), 119.7 (CH), 122.5 (C), 123.3 (C), 124.4 (CH), 126.4 (CH), 126.8 (CH), 127.9 (C), 128.0 (CH), 128.3 (CH), 129.0 (CH), 129.7 (CH), 130.8 (CH), 131.0 (C). 132.2 (C), 132.6 (CH), 132.78 (CH), 132.82 (C), 133.3 (CH), 146.3 (CH), 154.5 (C). HRMS (ESI) m/z: 451.0804 calcd for C27H20BrN2+ [M + H]+, found 451.0821. IR (KBr, cm−1): ν 3498, 3130, 1632, 1436.
1-(2-(2-Bromophenyl)-1-methyl-4-phenyl-1H-pyrrol-3-yl)pyridin-1-ium iodide (5a). To a stirred suspension of 2-(2-bromophenyl)-4-phenyl-3-(pyridin-1-ium-1-yl)pyrrol-1-ide (4a) (300 mg, 0.8 mmol) and K2CO3 (220 mg, 1.6 mmol, 2 equiv.) in acetonitrile (5 mL) methyl iodide (1.136 g, 8 mmol, 10 equiv.) was added in one portion, and the reaction mixture was vigorously stirred at rt for 12 h. After the reaction was completed acetonitrile and excess of methyl iodide were evaporated and the residue was suspended in water. Precipitate was collected, washed with water (3 × 5 mL) and dried to obtain 5a as bright-yellow solid, mp 248–250 °C, yield 410 mg, 99%. 1H NMR (DMSO-d6): δ 3.54 (s, 3H), 7.06 (d, J = 7.3 Hz, 2H), 7.22–7.28 (m, 1H), 7.28–7.36 (m, 2H), 7.40–7.48 (m, 1H), 7.48–7.56 (m, 1H), 7.58 (s, 1H), 7.67 (d, J = 7.3 Hz, 1H), 7.74 (d, J = 7.9 Hz, 1H), 8.08–8.20 (m, 2H), 8.6–8.72 (m, 1H), 8.94 (d, J = 5.7 Hz, 2H). 13C NMR (DMSO-d6): δ 34.8 (CH3), 117.8 (C), 121.2 (CH), 123.4 (C), 125.0 (C), 126.6 (CH), 127.0 (CH), 127.9 (C), 128.3 (CH), 128.5 (CH), 128.6 (C), 129.2 (CH), 130.9 (C), 132.3 (CH), 132.9 (CH), 133.9 (CH), 146.8 (CH), 147.2 (CH). HRMS (ESI) m/z: 389.0648 calcd for C22H18BrN2+ [M − I]+, found 389.0652. IR (KBr, cm−1): ν 3413, 3056, 1622, 1602, 1468.
1-(4-(2-Bromophenyl)-1-methyl-2-phenyl-1H-pyrrol-3-yl)pyridin-1-ium iodide (5b). Bright-yellow solid, mp 238–239 °C, yield 279 mg, 72%, obtained from 4-(2-bromophenyl)-2-phenyl-3-(pyridin-1-ium-1-yl)pyrrol-1-ide (4b) (281 mg, 0.75 mmol), methyl iodide (1.065 g, 7.5 mmol, 10 equiv.) and K2CO3 (207 mg, 1.5 mmol, 2 equiv.) according to procedure for 5a.1H NMR (CDCl3): δ 3.67 (s, 3H), 6.91 (s, 1H), 7.15–7.22 (m, 1H), 7.33–7.50 (m, 7H), 7.56 (dd, J = 7.6 Hz, J = 1.3 Hz, 1H), 8.02–8.12 (m, 2H), 8.52–8.60 (m, 1H), 8.62 (d, J = 5.6 Hz, 2H). 13C NMR (DMSO-d6): δ 35.3 (CH3), 117.2 (C), 122.7 (CH), 123.4 (C), 123.9 (C), 126.9 (C), 128.2 (CH), 128.2 (CH), 128.7 (C), 129.2 (CH), 129.3 (CH), 130.0 (CH), 130.1 (CH), 131.6 (C), 132.8 (CH), 132.9 (CH), 146.6 (CH), 146.7 (CH). HRMS (ESI) m/z: 389.0648 calcd for C22H18BrN2+ [M − I]+, found 389.0653. IR (KBr, cm−1): ν 3434, 3049, 1623, 1465.
1-(1-Acetyl-2-(2-bromophenyl)-4-phenyl-1H-pyrrol-3-yl)pyridin-1-ium chloride (6). To a stirred suspension of 2-(2-bromophenyl)-4-phenyl-3-(pyridin-1-ium-1-yl)pyrrol-1-ide (4a) (500 mg, 1.33 mmol) and K2CO3 (551 mg, 3.99 mmol, 3 equiv.) in dry DCM (30 mL) acetyl chloride (125 mg, 1.6 mmol, 1.2 equiv.) was added in one portion, and the reaction mixture was vigorously stirred at rt for 10 min (it is important not to stir longer than 10 min). Then reaction mixture was washed with brine (3 × 15 mL), dried under Na2SO4 and evaporated to dryness to obtain 6 as colorless solid, mp 300 °C, yield 597 mg, 99%. 1H NMR (CDCl3): δ 2.40 (s, 3H), 7.13 (dd, J = 6.6 Hz, J = 3.0 Hz, 2H), 7.26–7.34 (m, 4H), 7.44–7.51 (m, 1H), 7.56 (dd, J = 8.1 Hz, J = 0.8 Hz, 1H), 7.72 (s, 1H), 8.00 (dd, J = 7.6 Hz, J = 1.5 Hz, 1H), 8.23–8.32 (m, 2H), 8.66–8.73 (m, 1H), 9.15 (br s, 2H). 13C NMR (CDCl3): δ 23.9 (CH3), 118.3 (CH), 122.3 (C), 124.7 (C), 127.5 (CH), 128.4 (C), 128.6 (CH), 128.8 (C), 129.0 (C), 129.0 (C), 129.1 (CH), 129.4 (CH), 132.0 (CH), 132.6 (CH), 133.5 (CH), 146.4 (CH), 147.7 (CH), 147.7 (CH), 167.5 (C). HRMS (ESI) m/z: 419.0577 calcd for C23H18BrN2O+ [M − Cl]+, found 419.0590. IR (KBr, cm−1): ν 3401, 3065, 3007, 1737.
General procedure for the synthesis of 1-(1-benzyl-1H-pyrrol-3-yl)pyridin-1-ium bromides 7a–c
To a stirred suspension of 3-(pyridin-1-ium-1-yl)pyrrol-1-ides 4 (3 mmol) and K2CO3 (828 mg, 6 mmol, 2 equiv.) in acetonitrile (5 mL) benzyl bromide (564 mg, 3.3 mmol, 1.1 equiv.) was added in one portion, and the reaction mixture was vigorously stirred at rt for 12 h. After the reaction was completed acetonitrile was evaporated and the residue was suspended in diethyl ether. Precipitate was collected, washed with diethyl ether (3 × 10 mL) and water (3 × 5 mL) and dried to obtain 7.
1-(1-Benzyl-2-(2-bromophenyl)-4-phenyl-1H-pyrrol-3-yl)pyridin-1-ium bromide (7a). Bright-yellow solid, mp 269–270 °C, yield 1.590 g, 97%, obtained from 2-(2-bromophenyl)-4-phenyl-3-(pyridin-1-ium-1-yl)pyrrol-1-ide (4a) (1.126 g, 3 mmol), benzyl bromide (564 mg, 3.3 mmol, 1.1 equiv.) and K2CO3 (828 mg, 6 mmol, 2 equiv.). 1H NMR (DMSO-d6): δ 4.99 (d, J = 15.4 Hz, 1H), 5.19 (d, J = 15.5 Hz, 1H), 6.99–7.12 (m, 4H), 7.21–7.35 (m, 6H), 7.35–7.43 (m, 1H), 7.43–7.49 (m, 1H), 7.58 (d, J = 7.2 Hz, 1H), 7.64 (d, J = 7.9 Hz, 1H), 7.74 (s, 1H), 8.07–8.17 (m, 2H), 8.6–8.70 (m, 1H), 8.98 (d, J = 5.8 Hz, 2H). 13C NMR (DMSO-d6): δ 51.2 (CH2), 118.1 (C), 120.9 (CH), 123.8 (C), 125.1 (C), 126.6 (CH), 127.1 (CH), 127.4 (CH), 127.7 (C), 127.8 (CH), 128.1 (CH), 128.4 (C), 128.45 (CH), 128.5 (CH), 129.2 (CH), 130.8 (C), 132.3 (CH), 132.8 (CH), 134.3 (CH), 136.4 (C), 146.9 (CH), 147.3 (CH). HRMS (ESI) m/z: 465.0961 calcd for C28H22BrN2+ [M − Br]+, found 465.0974. IR (KBr, cm−1): ν 3025, 1623, 1604, 1466, 1450, 771.
1-(1-Benzyl-2-(2-bromophenyl)-4,5-diphenyl-1H-pyrrol-3-yl)pyridin-1-ium bromide (7b). Bright-yellow solid, mp 98–100 °C, yield 1.138 g, 61%, obtained from 2-(2-bromophenyl)-4,5-diphenyl-3-(pyridin-1-ium-1-yl)pyrrol-1-ide (4c) (1.354 g, 3 mmol), benzyl bromide (564 mg, 3.3 mmol, 1.1 equiv.) and K2CO3 (828 mg, 6 mmol, 2 equiv.). After work up described in general procedure product (Rf 0.15, DCM/MeOH 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was additionally purified by column chromatography on silica gel (DCM/MeOH from 50:1 to 1:1). 1H NMR (DMSO-d6): δ 4.90 (d, J = 16.7 Hz, 1H), 5.19 (d, J = 16.7 Hz, 1H), 6.81 (dd, J = 6.5 Hz, J = 2.8 Hz, 2H), 6.99 (dd, J = 6.5 Hz, J = 3.2 Hz, 2H), 7.14–7.23 (m, 6H), 7.28–7.33 (m, 2H), 7.33–7.46 (m, 5H), 7.64 (dd, J = 8.0 Hz, J = 1.1 Hz, 1H), 7.68 (dd, J = 7.6 Hz, J = 1.7 Hz, 1H), 8.08–8.15 (m, 2H), 8.59–8.67 (m, 1H), 9.01 (d, J = 5.6 Hz, 2H). 13C NMR (DMSO-d6): δ 48.4 (CH2), 117.7 (C), 125.0 (C), 125.03 (C), 126.1 (CH), 127.2 (CH), 127.3 (CH), 127.7 (C), 127.8 (C), 128.1 (CH), 128.2 (CH), 128.3 (CH), 128.6 (CH), 128.64 (CH), 128.8 (CH), 129.4 (CH), 129.8 (C), 130.4 (C), 131.0 (CH), 131.7 (C), 132.2 (CH), 132.8 (CH), 134.2 (CH), 136.6 (C), 146.9 (CH), 147.2 (CH). HRMS (ESI) m/z: 541.1274 calcd for C34H26BrN2+ [M − Br]+, found 541.1285. IR (KBr, cm−1): ν 3393, 3028, 1623, 1469, 1351, 762, 702.
:1) was additionally purified by column chromatography on silica gel (DCM/MeOH from 50:1 to 1:1). 1H NMR (DMSO-d6): δ 4.90 (d, J = 16.7 Hz, 1H), 5.19 (d, J = 16.7 Hz, 1H), 6.81 (dd, J = 6.5 Hz, J = 2.8 Hz, 2H), 6.99 (dd, J = 6.5 Hz, J = 3.2 Hz, 2H), 7.14–7.23 (m, 6H), 7.28–7.33 (m, 2H), 7.33–7.46 (m, 5H), 7.64 (dd, J = 8.0 Hz, J = 1.1 Hz, 1H), 7.68 (dd, J = 7.6 Hz, J = 1.7 Hz, 1H), 8.08–8.15 (m, 2H), 8.59–8.67 (m, 1H), 9.01 (d, J = 5.6 Hz, 2H). 13C NMR (DMSO-d6): δ 48.4 (CH2), 117.7 (C), 125.0 (C), 125.03 (C), 126.1 (CH), 127.2 (CH), 127.3 (CH), 127.7 (C), 127.8 (C), 128.1 (CH), 128.2 (CH), 128.3 (CH), 128.6 (CH), 128.64 (CH), 128.8 (CH), 129.4 (CH), 129.8 (C), 130.4 (C), 131.0 (CH), 131.7 (C), 132.2 (CH), 132.8 (CH), 134.2 (CH), 136.6 (C), 146.9 (CH), 147.2 (CH). HRMS (ESI) m/z: 541.1274 calcd for C34H26BrN2+ [M − Br]+, found 541.1285. IR (KBr, cm−1): ν 3393, 3028, 1623, 1469, 1351, 762, 702.
1-(1-Benzyl-2-(2-bromophenyl)-4-phenyl-1H-pyrrol-3-yl)-4-phenylpyridin-1-ium bromide (7c). Dark-yellow solid, mp 220–221 °C, yield 1.848 g, 99%, obtained from 2-(2-bromophenyl)-4-phenyl-3-(4-phenylpyridin-1-ium-1-yl)pyrrol-1-ide (4d) (1.354 g, 3 mmol), benzyl bromide (564 mg, 3.3 mmol, 1.1 equiv.) and K2CO3 (828 mg, 6 mmol, 2 equiv.). 1H NMR (DMSO-d6): δ 4.99 (d, J = 15.4 Hz, 1H), 5.20 (d, J = 15.5 Hz, 1H), 7.0–7.10 (m, 2H), 7.15 (d, J = 7.2 Hz, 2H), 7.22–7.37 (m, 6H), 7.37–7.44 (m, 1H), 7.44–7.50 (m, 1H), 7.54–7.70 (m, 5H), 7.74 (s, 1H), 8.10 (d, J = 7.4 Hz, 2H), 8.51 (d, J = 6.9 Hz, 2H), 8.97 (d, J = 6.8 Hz, 2H). 13C NMR (DMSO-d6): δ 51.1 (CH2), 118.3 (C), 120.3 (CH), 123.3 (C), 124.3 (CH), 125.2 (C), 126.7 (CH), 127.0 (CH), 127.4 (CH), 127.8 (CH), 127.9 (C), 128.1 (CH), 128.4 (C), 128.45 (CH), 128.5 (CH), 129.2 (CH), 129.7 (CH), 131.0 (C), 132.2 (CH), 132.6 (C), 132.79 (CH), 132.82 (CH), 134.3 (CH), 136.5 (C), 146.6 (CH), 155.5 (C). HRMS (ESI) m/z: 541.1274 calcd for C34H26BrN2+ [M − Br]+, found 541.1282. IR (KBr, cm−1): ν 3404, 3048, 1630.
1-Methyl-3-phenyl-1H-pyrido[2,1-a]pyrrolo[3,2-c]isoquinolin-4-ium iodide (8). 1-(2-(2-Bromophenyl)-1-methyl-4-phenyl-1H-pyrrol-3-yl)pyridin-1-ium iodide (5a) (100 mg, 0.193 mmol), K2CO3 (133 mg, 0.965 mmol, 5 equiv.), LiCl (12 mg, 0.290 mmol, 1.5 equiv.), TBAB (41 mg, 0.127 mmol, 0.66 equiv.) and 35 mL of DMF were placed in a flask with screw-cap. Argon was bubbled through this suspension and palladium(II) acetate (9 mg, 0.0386 mmol, 0.20 equiv.) was added. Argon was bubbled through reaction mixture again, flask was tightly screwed. Reaction mixture was vigorously stirred for 20 h at 60 °C (temperature inside oil bath). Then DMF was evaporated under reduced pressure. The solid residue was extracted with water (100 mL). Solid was filtered off and washed with water (50 mL). Combined water fractions were evaporated to 4 mL volume under reduced pressure. The solid precipitated from this solution was filtered, washed with cold water (2 mL) and dried to obtain pure 8 as bright-yellow crystals, mp 262–263 °C, yield 24 mg, 29%. 1H NMR (DMSO-d6): δ 4.46 (s, 3H), 7.53–7.64 (m, 5H), 7.88 (s, 1H), 7.93–7.99 (m, 1H), 7.99–8.06 (m, 1H), 8.15–8.22 (m, 1H), 8.45–8.52 (m, 1H), 8.80 (d, J = 8.4 Hz, 1H), 9.16 (d, J = 6.7 Hz, 1H), 9.24 (d, J = 8.5 Hz, 1H), 9.57 (d, J = 8.8 Hz, 1H). 13C NMR (DMSO-d6): δ 38.9 (CH3), 113.2 (C), 121.2 (C), 121.5 (CH), 121.8 (C), 123.5 (C), 123.54 (CH), 124.2 (CH), 124.7 (C), 126.9 (CH), 127.6 (CH), 128.4 (CH), 129.2 (CH), 130.1 (CH), 132.4 (CH), 132.6 (C), 133.4 (CH), 133.8 (CH), 137.2 (CH), 140.0 (C). HRMS (ESI) m/z: 309.1386 calcd for C22H17N2+ [M − I]+, found 309.1388. IR (KBr, cm−1): ν 3436, 3039, 1614, 1463.
1-Benzyl-3-phenyl-1H-pyrido[2,1-a]pyrrolo[3,2-c]isoquinolin-4-ium bromide (9a). 1-(1-Benzyl-2-(2-bromophenyl)-4-phenyl-1H-pyrrol-3-yl)pyridin-1-ium bromide (7a) (1.500 g, 2.75 mmol), K2CO3 (1.898 g, 13.75 mmol, 5 equiv.), LiCl (175 mg, 4.13 mmol, 1.5 equiv.), TBAB (585 mg, 1.82 mmol, 0.66 equiv.) and 200 mL of DMF were placed in a flask with screw-cap. Argon was bubbled through this suspension and palladium(II) acetate (123 mg, 0.55 mmol, 0.20 equiv.) was added. Argon was bubbled through reaction mixture again, flask was tightly screwed. Reaction mixture was vigorously stirred for 30 h at 60 °C (temperature inside oil bath). Then DMF was evaporated under reduced pressure. The solid residue was extracted with water (3 × 350 mL). Every time after extraction the solid residue was filtered off and new portion of water was added to it. Combined water phases were evaporated to 20 mL volume under reduced pressure. The solid precipitated from this solution was filtered, washed with cold water (5 mL) and dried to obtain pure 9a as bright-yellow crystals, mp 107–110 °C, yield 746 mg, 58%. 1H NMR (CDCl3): δ 6.02 (s, 2H), 7.20 (d, J = 7.0 Hz, 2H), 7.31–7.42 (m, 3H), 7.44 (s, 1H), 7.49–7.62 (m, 5H), 7.80–7.93 (m, 3H), 8.27 (dd, J = 6.8 Hz, J = 2.5 Hz, 1H), 8.72–8.80 (m, 1H), 9.25 (d, J = 6.6 Hz, 1H), 9.32–9.39 (m, 1H), 9.98 (d, J = 8.9 Hz, 1H). 13C NMR (CDCl3): δ 54.9 (CH2), 114.9 (C), 121.3 (CH), 121.6 (C), 122.3 (C), 123.9 (CH), 124.1 (C), 124.4 (C), 126.2 (CH), 126.23 (CH), 128.2 (CH), 128.6 (CH), 128.7 (CH), 129.3 (CH), 129.5 (CH), 129.7 (CH), 130.4 (CH), 131.3 (CH), 131.8 (C), 132.6 (CH), 134.3 (CH), 134.7 (C), 138.8 (CH), 140.6 (C). HRMS (ESI) m/z: 385.1699 calcd for C28H21N2+ [M − Br]+, found 385.1716. IR (KBr, cm−1): ν 3382, 3056, 1614, 1463, 1417.
1-Benzyl-2,3-diphenyl-1H-pyrido[2,1-a]pyrrolo[3,2-c]isoquinolin-4-ium bromide (9b). 1-(1-Benzyl-2-(2-bromophenyl)-4,5-diphenyl-1H-pyrrol-3-yl)pyridin-1-ium bromide (7b) (300 mg, 0.482 mmol), K2CO3 (333 mg, 2.41 mmol, 5 equiv.), LiCl (31 mg, 0.723 mmol, 1.5 equiv.), TBAB (103 mg, 0.318 mmol, 0.66 equiv.) and 50 mL of DMF were placed in a flask with screw-cap. Argon was bubbled through this suspension and palladium(II) acetate (22 mg, 0.964 mmol, 0.20 equiv.) was added. Argon was bubbled through reaction mixture again, flask was tightly screwed. Reaction mixture was vigorously stirred for 47 h at 70 °C (temperature inside oil bath). Then DMF was evaporated under reduced pressure. The solid residue was extracted with water (3 × 250 mL). Every time after extraction the solid residue was filtered off and new portion of water was added to it. Combined water phases were evaporated to 5 mL volume under reduced pressure. The solid precipitated from this solution was filtered, washed with cold water (5 mL) and dried to obtain pure 9b as bright-yellow crystals, mp 272 °C, yield 108 mg, 41%. 1H NMR (DMSO-d6): δ 5.88 (s, 2H), 7.10 (d, J = 7.3 Hz, 2H), 7.24–7.41 (m, 8H), 7.45–7.54 (m, 5H), 7.84–7.91 (m, 1H), 9.93–8.03 (m, 2H), 8.39 (d, J = 8.4 Hz, 1H), 8.46–8.56 (m, 1H), 9.06 (d, J = 6.6 Hz, 1H), 9.25 (d, J = 8.4 Hz, 1H), 9.61 (d, J = 8.8 Hz, 1H). 13C NMR (DMSO-d6): δ 50.3 (CH2), 113.4 (C), 121.7 (CH), 122.1 (C), 122.4 (C), 122.8 (C), 123.5 (CH), 123.8 (C), 124.3 (CH), 125.4 (CH), 127.1 (CH), 127.6 (CH), 127.6 (CH), 128.4 (CH), 128.5 (CH), 128.8 (C), 129.0 (CH), 129.1 (CH), 129.4 (CH), 130.9 (CH), 131.3 (CH), 132.4 (C), 133.3 (CH), 133.7 (CH), 136.4 (C), 137.5 (CH), 140.3 (C), 141.6 (C). HRMS (ESI) m/z: 461.2013 calcd for C34H25N2+ [M − Br]+, found 461.2057. IR (KBr, cm−1): ν 3438, 1614, 1466, 1419.
1-Benzyl-3,7-diphenyl-1H-pyrido[2,1-a]pyrrolo[3,2-c]isoquinolin-4-ium bromide (9c). 1-(1-Benzyl-2-(2-bromophenyl)-4-phenyl-1H-pyrrol-3-yl)-4-phenylpyridin-1-ium bromide (7c) (1.500 g, 2.41 mmol), K2CO3 (1.663 g, 12.05 mmol, 5 equiv.), LiCl (153 mg, 3.62 mmol, 1.5 equiv.), TBAB (513 mg, 1.59 mmol, 0.66 equiv.) and 200 mL of DMF were placed in a flask with screw-cap. Argon was bubbled through this suspension and palladium(II) acetate (108 mg, 0.482 mmol, 0.20 equiv.) was added. Argon was bubbled through reaction mixture again, flask was tightly screwed. Reaction mixture was vigorously stirred for 30 h at 60 °C (temperature inside oil bath). Then DMF was evaporated under reduced pressure. The solid residue was washed with water and dried. Then it was treated with methanol (250 mL) and suspension obtained was filtered. Filtrate (methanol solution of the product) was evaporated under reduced pressure. The solid residue was treated with refluxing ethyl acetate (100 mL) for 30 min. Then it was allowed to cool to rt. The solid was filtered, washed with ethyl acetate and dried to obtain pure 9c as orange-yellow crystals, mp 183 °C, yield 733 mg, 56%. 1H NMR (DMSO-d6): δ 6.15 (s, 2H), 7.22 (d, J = 7.4 Hz, 2H), 7.26–7.32 (m, 1H), 7.33–7.41 (m, 2H), 7.56–7.75 (m, 8H), 7.83–7.91 (m, 1H), 7.96–8.03 (m, 1H), 8.07 (s, 1H), 8.23–8.31 (m, 2H), 8.42 (d, J = 8.4 Hz, 1H), 8.51 (dd, J = 7.3 Hz, J = 1.8 Hz, 1H), 9.22 (d, J = 7.3 Hz, 1H), 9.51 (d, J = 8.5 Hz, 1H), 9.70 (s, 1H). 13C NMR (DMSO-d6): δ 53.4 (CH2), 113.9 (C), 119.7 (CH), 120.9 (CH), 121.5 (CH), 121.7 (C), 122.2 (C), 122.9 (C), 124.2 (C), 126.0 (CH), 127.5 (CH), 127.6 (CH), 127.8 (CH), 128.1 (CH), 128.5 (CH), 129.0 (CH), 129.3 (CH), 129.5 (CH), 130.3 (CH), 131.3 (CH), 132.5 (C), 132.6 (CH), 133.6 (CH), 133.7 (CH), 134.2 (C), 136.5 (C), 140.5 (C), 147.2 (C). HRMS (ESI) m/z: 461.2012 calcd for C34H25N2+ [M − Br]+, found 461.2021. IR (KBr, cm−1): ν 3392, 3052, 1635, 1612.
Debenzylation of 1-benzyl-3-phenyl-1H-pyrido[2,1-a]pyrrolo[3,2-c]isoquinolin-4-ium bromide (9a) with hydrogen. Suspension of 9a (15 mg, 0.0322 mmol) and Adams catalyst (2 mg, 13 wt%) in methanol (5 mL) was stirred at 60 °C for 12 h under pressure of balloon with hydrogen. Then reaction mixture was filtered, evaporated to dryness. Column chromatography on silica gel (DCM/MeOH 20:1 to 10:1) allowed to obtain 1-benzyl-3-phenyl-5,6,7,8-tetrahydro-1H-pyrido[2,1-a]pyrrolo[3,2-c]isoquinolin-4-ium bromide (10) (Rf 0.15, DCM/MeOH 10:1) as yellowish solid, yield 4 mg, 27%. 1H NMR (CDCl3): δ 2.06–2.14 (m, 2H), 2.26–2.35 (m, 2H), 4.04 (t, J = 6.5 Hz, 2H), 4.67 (t, J = 5.9 Hz, 2H), 5.94 (s, 2H), 7.17 (d, J = 7.1 Hz, 2H), 7.33–7.44 (m, 4H), 7.47–7.53 (m, 3H), 7.60–7.65 (m, 2H), 7.44–7.80 (m, 1H), 7.89–7.96 (m, 1H), 8.26 (d, J = 8.6 Hz, 1H), 8.60 (d, J = 8.6 Hz, 1H). 13C NMR (CDCl3): δ 17.8 (CH2), 21.2 (CH2), 27.4 (CH2), 29.7 (CH2), 54.5 (CH2), 114.8 (C), 121.3 (CH), 123.6 (C), 124.8 (C), 126.3 (CH), 126.7 (C), 127.7 (CH), 128.1 (C), 128.5 (CH), 128.59 (CH), 128.60 (CH), 128.65 (CH), 129.5 (CH), 131.2 (CH), 132.7 (CH), 133.0 (C), 134.8 (C), 135.3 (CH), 154.2 (C). HRMS (ESI) m/z: 389.2012 calcd for C28H25N2+ [M − Br]+, found 389.2019.
2-Phenylpyrido[2,1-a]pyrrolo[3,2-c]isoquinoline (11). A suspension of 1-benzyl-3-phenyl-1H-pyrido[2,1-a]pyrrolo[3,2-c]isoquinolin-4-ium bromide (9a) (700 mg, 1.504 mmol) and AlCl3 (1.004 g, 7.52 mmol, 5 equiv.) in benzene (20 mL) was refluxed for 3 h. Then reaction mixture was evaporated to dryness under reduced pressure. The solid residue was dissolved in sufficient for complete solution amount of aq. 3 wt% solution of HCl (up to 1 L). This acidic solution was washed with diethyl ether (3 × 200 mL), made basic with aq. 10 wt% solution of KOH and extracted with DCM (4 × 200 mL). Combined DCM phases were dried under Na2SO4, filtered and evaporated under reduced pressure to obtain 11 as cherry red solid, mp 155–158 °C, yield 440 mg, 99%. 1H NMR (CDCl3): δ 7.12 (s, 1H), 7.23–7.30 (m, 1H), 7.38–7.48 (m, 4H), 7.56–7.65 (m, 1H), 7.73–7.82 (m, 1H), 8.15 (d, J = 7.4 Hz, 2H), 8.32 (d, J = 8.6 Hz, 1H), 8.58 (d, J = 8.8 Hz, 1H), 8.71 (d, J = 6.4 Hz, 1H), 8.91 (d, J = 8.3 Hz, 1H). 13C NMR (CDCl3): δ 90.6 (CH), 118.6 (C), 120.7 (CH), 122.2 (CH), 122.6 (CH), 123.9 (CH), 124.6 (CH), 126.1 (CH), 126.8 (CH), 128.2 (CH), 128.5 (CH), 128.7 (C), 129.5 (C), 130.6 (CH), 131.5 (CH), 135.0 (C), 135.4 (C), 137.3 (C), 151.5 (C). HRMS (ESI) m/z: 295.1230 calcd for C21H15N2+ [M + H]+, found 295.1240. IR (KBr, cm−1): ν 3365, 1473, 1378.
2,3-Diphenylpyrido[2,1-a]pyrrolo[3,2-c]isoquinoline (12). A suspension of 1-benzyl-2,3-diphenyl-1H-pyrido[2,1-a]pyrrolo[3,2-c]isoquinolin-4-ium bromide (9b) (100 mg, 0.185 mmol) and AlCl3 (123 mg, 0.923 mmol, 5 equiv.) in benzene (5 mL) was refluxed for 3 h. The work up procedure is similar to protocol for 12. Compound 12 is cherry-red solid, mp 214 °C, yield 68 mg, 99%. 1H NMR (CDCl3): δ 7.12–7.18 (m, 1H), 7.19–7.28 (m, 3H), 7.41–7.51 (m, 5H), 7.52–7.58 (m, 1H), 7.66 (d, J = 7.2 Hz, 2H), 7.68–7.75 (m, 1H), 7.82–7.89 (m, 1H), 8.51 (d, J = 8.6 Hz, 1H), 8.84 (d, J = 8.8 Hz, 1H), 9.02 (d, J = 6.9 Hz, 1H), 9.15 (d, J = 7.9 Hz, 1H). 13C NMR (CDCl3): δ 110.1 (C), 118.7 (C), 119.9 (CH), 122.6 (CH), 122.9 (CH), 123.8 (CH), 124.7 (CH), 124.9 (C), 126.1 (CH), 127.1 (CH), 127.8 (CH), 128.1 (CH), 128.9 (CH), 129.1 (CH), 130.3 (C), 130.6 (CH), 131.6 (CH), 132.1 (CH), 135.2 (C), 135.9 (C), 137.8 (C), 138.1 (C), 150.1 (C). HRMS (ESI) m/z: 371.1543 calcd for C27H19N2+ [M + H]+, found 371.1557. IR (KBr, cm−1): ν 3416, 3046, 1380.
2,7-Diphenylpyrido[2,1-a]pyrrolo[3,2-c]isoquinoline (13). A suspension of 1-benzyl-3,7-diphenyl-1H-pyrido[2,1-a]pyrrolo[3,2-c]isoquinolin-4-ium bromide (9c) (500 mg, 0.923 mmol) and AlCl3 (616 mg, 4.62 mmol, 5 equiv.) in benzene (20 mL) was refluxed for 7 h. Then reaction mixture was evaporated to dryness under reduced pressure. The solid residue was suspended in aq. 5 wt% solution of HCl (100 mL) and filtered. Precipitate was washed with water (50 mL) and diethyl ether (150 mL) and dissolved in a mixture of aq. 10 wt% solution of KOH (200 mL) and DCM (200 mL). DCM layer was separated and basic layer was extracted with DCM (2 × 200 mL). Combined DCM phases were dried under Na2SO4, filtered and evaporated under reduced pressure to obtain 13 as dark red solid, mp 260 °C, yield 340 mg, 99%. 1H NMR (CDCl3): δ 7.26–7.31 (m, 2H), 7.41–7.64 (m, 6H), 7.72–7.85 (m, 4H), 8.23 (d, J = 7.3 Hz, 2H), 8.50 (d, J = 8.6 Hz, 1H), 8.81–8.87 (m, 1H), 8.85 (s, 1H), 8.93 (d, J = 8.2 Hz, 1H). 13C NMR (CDCl3): δ 90.5 (CH), 118.9 (CH), 119.1 (C), 119.3 (CH), 122.8 (CH), 123.9 (CH), 124.5 (CH), 126.1 (CH), 126.7 (CH), 127.0 (CH), 128.5 (CH), 128.7 (C), 129.6 (CH), 129.8 (CH), 130.1 (C), 130.8 (CH), 131.4 (CH), 135.0 (C), 136.2 (C), 136.6 (C), 137.7 (C), 140.3 (C), 152.3 (C). HRMS (ESI) m/z: 371.1543 calcd for C27H19N2+ [M + H]+, found 371.1561. IR (KBr, cm−1): ν 3051, 1377, 1210.
Acknowledgements
We gratefully acknowledge the financial support of the Russian Foundation for Basic Research (Grant No. 14-03-00187) and Saint Petersburg State University (Grant No. 12.50.1565.2013, 12.38.239.2014, 12.38.217.2015). This research was carried out using resources of the X-ray Diffraction Centre, the Centre for Magnetic Resonance, the Computer Centre, the Centre for Optical and Laser Materials Research and the Centre for Chemical Analysis and Materials of St. Petersburg State University.
Notes and references
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, 3rd edn, 2006 Search PubMed.
- For recent examples see:
(a) E. Kim, Y. Lee, S. Lee and S. B. Park, Acc. Chem. Res., 2015, 48, 538–547 CrossRef CAS PubMed;
(b) D.-T. Yang, J. Radtke, S. K. Mellerup, K. Yuan, X. Wang, M. Wagner and S. Wang, Org. Lett., 2015, 17, 2486–2489 CrossRef CAS PubMed;
(c) G. Liu, D. Chen, L. Kong, J. Shi, B. Tong, J. Zhi, X. Feng and Y. Dong, Chem. Commun., 2015, 51, 8555–8558 RSC;
(d) I.-S. Tamgho, A. Hasheminasab, J. T. Engle, V. N. Nemykin and C. J. Ziegler, J. Am. Chem. Soc., 2014, 136, 5623–5626 CrossRef CAS PubMed;
(e) A. K. A. Almeida, M. P. Monteiro, J. M. M. Dias, L. Omena, A. J. C. da Silva, J. Tonholo, R. J. Mortimer, M. Navarro, C. Jacinto, A. S. Ribeiro and I. N. de Oliveira, Spectrochim. Acta, Part A, 2014, 128, 812–818 CrossRef CAS PubMed;
(f) L. He, W. Lin, Q. Xu and H. Wei, ACS Appl. Mater. Interfaces, 2014, 6, 22326–22333 CrossRef CAS PubMed;
(g) J. Bhosale, U. Fegade, B. Bondhopadhyay, S. Kaur, N. Singh, A. Basu, R. Dabur, R. Bendre and A. Kuwar, J. Mol. Recognit., 2015, 28, 369–375 CrossRef CAS PubMed;
(h) M. Sokhanvar and M. Pordel, ARKIVOC, 2014, iv, 328–341 Search PubMed;
(i) B. Liu, Z. Wang, N. Wu, M. Li, J. You and J. Lan, Chem.–Eur. J., 2012, 18, 1599–1603 CrossRef CAS PubMed.
- For recent publications see:
(a) E. E. Galenko, O. A. Tomashenko, A. F. Khlebnikov and M. S. Novikov, Org. Biomol. Chem., 2015, 13, 9825–9833 RSC;
(b) A. V. Galenko, A. F. Khlebnikov, M. S. Novikov and M. S. Avdontseva, Tetrahedron, 2015, 71, 1940–1951 CrossRef CAS;
(c) E. E. Galenko, A. V. Galenko, A. F. Khlebnikov and M. S. Novikov, RSC Adv., 2015, 5, 18172–18176 RSC;
(d) M. S. Novikov, A. F. Khlebnikov, N. V. Rostovskii, S. Tcyrulnikov, A. A. Suhanova, K. V. Zavyalov and D. S. Yufit, J. Org. Chem., 2015, 80, 18–29 CrossRef CAS PubMed;
(e) A. F. Khlebnikov, O. A. Tomashenko, L. D. Funt and M. S. Novikov, Org. Biomol. Chem., 2014, 12, 6598–6609 RSC;
(f) K. V. Zavyalov, M. S. Novikov, A. F. Khlebnikov and V. V. Pakalnis, Tetrahedron, 2014, 70, 3377–3384 CrossRef CAS;
(g) A. S. Konev, A. F. Khlebnikov, T. G. Nikiforova, A. A. Virtsev and H. Frauendorf, J. Org. Chem., 2013, 78, 2542–2552 CrossRef CAS PubMed;
(h) A. F. Khlebnikov, M. V. Golovkina, M. S. Novikov and D. S. Yufit, Org. Lett., 2012, 14, 3768–3771 CrossRef CAS PubMed.
-
(a) J. A. Murphy and M. S. Sherburn, Tetrahedron Lett., 1990, 31, 3495–3496 CrossRef CAS;
(b) R. R. Castillo, C. Burgos, J. J. Vaquero and J. Alvarez-Builla, Eur. J. Org. Chem., 2011, 619–628 CrossRef CAS.
- S. Hernandez, R. SanMartin, I. Tellitu and E. Dominguez, Org. Lett., 2003, 5, 1095–1098 CrossRef CAS PubMed.
-
(a) A. Furstner, M. M. Domostoj and B. Scheiper, J. Am. Chem. Soc., 2006, 128, 8087–8094 CrossRef PubMed;
(b) M. Egorov, B. Delpech, G. Aubert, T. Cresteil, M. C. Garcia-Alvarez, P. Collin and C. Marazano, Org. Biomol. Chem., 2014, 12, 1518–1524 RSC.
-
(a) R. M. Suárez, P. Bosch, D. Sucunza, A. M. Cuadro, A. Domingo, F. Mendicutic and J. J. Vaquero, Org. Biomol. Chem., 2015, 13, 527–538 RSC;
(b) G. Marcelo, S. Pinto, T. Cañeque, I. F. A. Mariz, A. M. Cuadro, J. J. Vaquero, J. M. G. Martinho and E. M. S. Maçôas, J. Phys. Chem. A, 2015, 119, 2351–2362 CrossRef CAS PubMed;
(c) K. Faulhaber, A. Granzhan, H. Ihmels, D. Otto, L. Thomas and S. Wells, Photochem. Photobiol. Sci., 2011, 10, 1535–1545 RSC;
(d) Zh. Chen, Sh. Zhang, X. Qi, Sh. Liu, Q. Zhanga and Y. Deng, J. Mater. Chem., 2011, 21, 8979–8982 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra for all new compounds, X-ray data, photophysical data and computational details. CCDC 1407107, 1407108, 1407277 and 1412486. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra17755c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.