
Effects of an acid–alkaline environment on the reactivity of 5-carboxycytosine with hydroxyl radicals†
Lingxia Jin*a,
Caibin Zhaoa,
Tianlei Zhanga,
Zhiyin Wanga,
Suotian Mina,
Wenliang Wang*b and
Yawen Weic
aShaanxi Province Key Laboratory of Catalytic Fundamentals & Applications, School of Chemical & Environment Science, Shaanxi University of Technology, Hanzhong, Shaanxi 723001, China. E-mail: jinlx@snut.edu.cn; wlwang@snnu.edu.cn; Fax: +86-29-81530727; Tel: +86-916-2641660, +86-29-81530815
bKey Laboratory for Macromolecular Science of Shaanxi Province, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi’an 710062, China
cInstitute of Publication Science, Chang’an University, Xi’an 710064, China
First published on 24th September 2015
Abstract
A hydroxyl radical (˙OH) is produced in biological systems by external or endogenous agents. It can damage DNA/RNA by attacking pyrimidine nucleobases through an addition reaction and H-atom abstraction. However, the correlation study for the new cytosine derived DNA modification (5-carboxycytosine, 5-caCyt) remains scarcely existent. Here three distinct groups of mechanisms for 5-caCyt with ˙OH by the CBS-QB3 approach have firstly been explored: the direct reaction (paths R1–R6), acidic (paths R1′–R3′, R5′, R6′), and alkaline (paths R1′′–R5′′)-induced processes. It indicates that the addition of ˙OH to the C5![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C6 double bond of 5-caCyt is more favourable in neutral, acidic and alkaline conditions, and the ΔGs≠ value of the C5 channel is a little higher than that of the C6 route, which agrees with the tendencies observed experimentally. Moreover, the H5 abstraction in alkaline media might be competitive with the addition reactions, having a ΔGs≠ value of 32.55 kJ mol−1, which is only 17–20 kJ mol−1 more energetic than for the addition reactions. In addition, the ΔGs≠ values of the ˙OH reactions are slightly lower for the neutral or deprotonated systems than for the N3-protonated 5-caCyt, implying that the reaction trends are a little enhanced. Our results give a possible new insight on 5-caCyt in the presence of ˙OH for experimental scientists.
C6 double bond of 5-caCyt is more favourable in neutral, acidic and alkaline conditions, and the ΔGs≠ value of the C5 channel is a little higher than that of the C6 route, which agrees with the tendencies observed experimentally. Moreover, the H5 abstraction in alkaline media might be competitive with the addition reactions, having a ΔGs≠ value of 32.55 kJ mol−1, which is only 17–20 kJ mol−1 more energetic than for the addition reactions. In addition, the ΔGs≠ values of the ˙OH reactions are slightly lower for the neutral or deprotonated systems than for the N3-protonated 5-caCyt, implying that the reaction trends are a little enhanced. Our results give a possible new insight on 5-caCyt in the presence of ˙OH for experimental scientists.
1. Introduction
The hydroxyl radical (˙OH) is an important ROS, which is usually present at very low levels in biological systems, mainly arising from oxygen metabolism. Its concentration is markedly enhanced upon exposure of cells to exogenous chemical and physical agents, such as ionizing radiation. Cells have mechanisms to palliate the toxicity of ˙OH when its concentration is low.1,2 However, at higher concentrations, damage is unavoidable and the development of age-dependent diseases such as cancer, arteriosclerosis, arthritis and neurodegenerative disorders are prevailing.3,4The recently discovered nucleobase 5-carboxylcytosine (5-caCyt) is the final product of oxidative attack on the C5 position of cytosine (Cyt) by TET proteins,5–8 which has been proposed as the eighth DNA base. It can be removed from DNA and replaced by Cyt via base excision repair.9 A study recently demonstrated that 5-caCyt in mouse embryonic stem cells may recruit unique proteins for certain functions; it could be more than the intermediates in the DNA demethylation pathway.10 Moreover, the experiments recently demonstrated that 5-caCyt can change the fidelity of DNA replication and slow down RNA polymerase II transcription, suggesting that the possible functional roles of 5-caCyt occur on DNA replication and transcription.11,12 On the contrary, it is still unclear whether 5-caCyt plays functional roles in cancer development and formation.13 According to the literature,14 the development of certain cancers is interconnected with oxidative damage by a hydroxyl radical. It can oxidize the DNA bases either by addition or hydrogen abstraction reactions to produce damaged bases or strand breaks.15,16 For instance, Zuo et al.17 discovered that the preferential addition of the OH radical to the C5 and C6 sites of 5-MeCyt would produce pyrimidine glycol, leading to the deamination of 5-MeCyt to thymine. Von Sonntag et al.18 demonstrated that hydrogen abstraction from the methyl group gave rise to the methyl radical of the pyrimidine bases. Those radicals may react with the neighboring nucleobases to cause damage to cell functions. Therefore, the reactions of ˙OH with 5-caCyt are also likely to happen and might also lead to damaged DNA bases that cause the formation of cancer. Moreover, the early experiments19 have reported that the adduct radicals are mainly formed by the addition of ˙OH to C5 and C6 sites of 5-caCyt in solution. As for 5-caCyt, it still possesses three double bonds and five H atoms, but what is to become of them in the face of ˙OH?
It has been demonstrated in experiments that there are three prevailing species of the 5-caCyt moiety in 5-carboxyl-2′-deoxycytidine (R = 2′-deoxyribosyl) at different pH, the cationic and anionic 5-caCyt, and the zwitterionic 5-caCyt± species.20 Portalone et al.21 have reported that only the anionic (b) and zwitterionic forms (c) have been detected in solution in the pH range 2.0–9.0 (Fig. 1). Unfortunately, the zwitterionic 5-caCyt± form fails to be optimized due to amino–imino conversion by means of theoretical calculation.21–23 The anionic form (5-CytCOO−) in the experimental studies has been artificially introduced in double-stranded DNA, and on the contrary the cationic form is not expected in cellular DNA at physiological conditions.20 The existence of the N3-protonated form (a) has been postulated at pH < 1 (Fig. S1 and Table S1†).20,21 However, at this strongly acidic pH, the DNA macromolecule should be hydrolysed,24 and the protonation of the N3 site for 5-caCyt (5-caCytN3+) could not exist in DNA in vivo. Therefore, the deprotonation of the C5-carboxylic group (5-CytCOO−) and neutral 5-caCyt will be considered, raising the question of whether there is competition between them when treated with ˙OH. Meanwhile, for contrast, the ˙OH-mediated 5-caCytN3+ reactions have also been reported in this paper. Moreover, until now the experimental data of these reactions in solution are vanishingly rare.
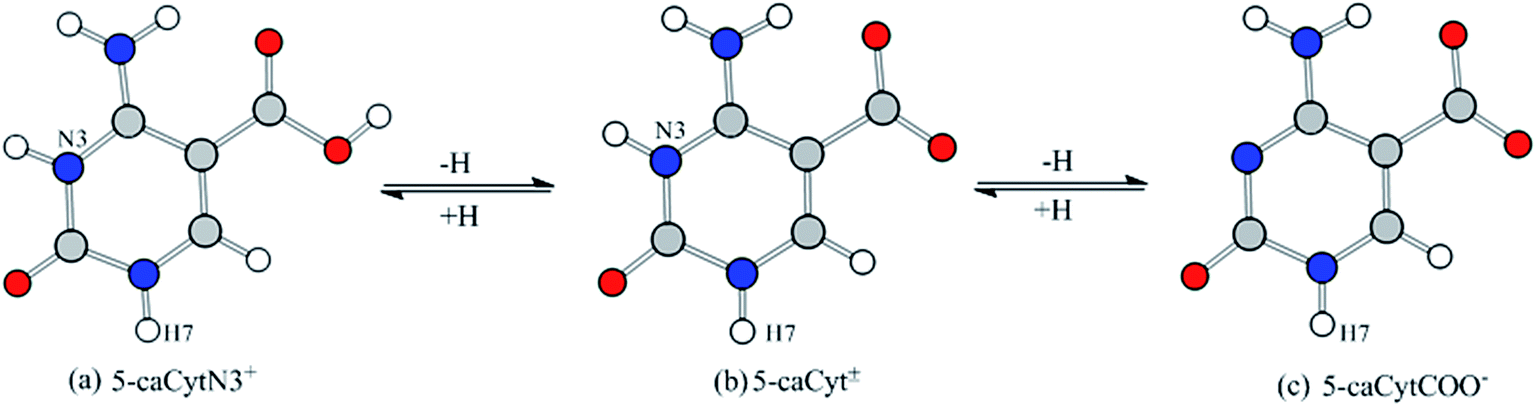 | ||
| Fig. 1 Prevailing species of the 5-caCyt moiety in 5-carboxyl-2′-deoxycytidine (H7 = 2′-deoxyribosyl) at different pH: (a) at pH < 1, (b) and (c) at pH = 2–9.21 | ||
As mentioned above, the ˙OH-mediated addition and H-atom abstraction mechanisms of the 5-caCyt and 5-CytCOO− are firstly explored from a theoretical perspective to clarify whether the ˙OH-mediated processes of 5-caCyt can kinetically compete with their 5-CytCOO−. Meanwhile, the reaction mechanisms and activation free energies of 5-caCyt and 5-CytCOO− have been performed and compared with their 5-caCytN3+ forms in the presence of ˙OH. Our results give a possible new insight into 5-caCytN3+ and 5-CytCOO− under abnormal environments such as hypoxia or hyperoxia.
2. Computational methods
All calculations were performed using the Gaussian 09 package.25 The more reliable energies were conducted at the CBS-QB3 level of theory.26 Specifically, the CBS-QB3 method involves the following steps: B3LYP/6-311G (2d,d,p) geometry optimization; B3LYP/6-311G (2d,d,p) frequency calculation with a 0.99 scale factor for the ZPE; CCSD (T)/6-31+G* energy calculation; MP4 (SDQ)/6-31+G(d(f),p) energy calculation; UMP2/6-311+G(3d2f,2df,2p) energy calculation and CBS extrapolation. In addition, this approach includes empirical corrections for spin contamination.To test the reliability of the CBS-QB3 method, single point computation was also done using the G3B3 approach.27 It is clear from Table S2† that the activation free energies calculated using both of the different methods agree well with each other, proving that these two approaches are able to provide reliable data for our system.
Moreover, many previous investigations have proposed that the CBS-QB3 method can provide adequately accurate energies, with a standard deviation of about 1.5 kcal mol−1.28–30 The CBS-QB3 method has also been used to calculate ΔG values for a deprotonation system where the experimental values are reported to be accurate within 1 kcal mol−1.31 We note that the CBS-QB3 methodology includes a correction for spin contamination in open-shell species.26,32–37 Therefore, the CBS-QB3 method was selected for radical additions to unsaturated bonds because these kinds of transition states suffer from spin contamination.
All stationary points including reactant complexes, transition states, and products have been optimized using the CBSB7 (B3LYP/6-311G (2d,d,p)) method. Vibrational frequency calculations have been conducted at the level of theory used for optimization to characterize the nature of each stationary point as a minimum (real frequencies) or transition state (only one imaginary frequency) and to correct energies for zero-point at 298.15 K. Intrinsic reaction coordinate (IRC)38 calculations have been carried out at the same level of theory from each transition state to ensure that the obtained transition states connected the appropriate reactants and products. Besides, the obtained stationary points based on the gas phase geometries have been further optimized using the polarized continuum model (PCM)39 at the CBS-QB3 method, with a dielectric constant of 78.39 to simulate the aqueous environment. Additionally, the activation free energies in the gas (ΔGg≠) and aqueous phase (ΔGs≠) calculations have been obtained at 298.15 K.
Finally, the nucleus-independent chemical shifts (NICS(0)) in the centers of the six-membered heterocyclic compounds of the lowest energy structures for the product radicals are determined by the GIAO/DFT procedure (the gauge-independent atomic orbital/density functional theory).40 The Natural Population Analysis (NPA) charges have been used to analyze the difference in the reaction trend in the gas and aqueous phases. They were calculated by the CBS-QB3 method,26 adopting natural population analysis.41,42
3. Results and discussion
The expectation values of 〈S2〉 for all species contained in these reaction systems are listed in Table S3.† After spin annihilation, the values of 〈S2〉 for the open-shell systems are very close to 0.7500, indicating that the spin contamination can be negligible at the above mentioned computation level.In neutral conditions, as for 5-caCyt, there are two isomers based on the torsion and angle of the COOH group, denoted as M1 and M2, respectively (Fig. S2 and Table S4†), and M1 both in the gas and aqueous phases are more stable than M2. Thus on the basis of this result, the more stable M1 isomer has been chosen for the present computational study. As seen from Fig. S2,† the dihedral angles are all 0.0° for the pyrimidine ring of 5-caCyt, suggesting a planar geometry in the ring π-system. The corresponding dihedral angles are also 0.0° for the exocyclic group of 5-caCyt, implying that the more planar character is found in CO, –NH2, and –COOH of 5-caCyt, respectively. The constituent atoms of these bonds are expected to be more reactive for the electrophilic addition reaction with a hydroxyl radical. The structural features of 5-caCyt favor C2, O2, N3, C4, C5, C6, C7, and O3 as the addition sites. For the O2 and O3 sites, various initial geometries of adducts have been designed, but the ˙OH is always far from O2 and O3 atoms. For the ˙OH addition to the C2, N3, C4, C5, C6 and C7 sites of 5-caCyt (Tables 1 and S5, Fig. S3†), the ΔEg≠ values between the initial reactants and the TSs are 58.51, 62.68, 45.11, 4.03, 1.07 and 63.10 kJ mol−1, respectively, suggesting that the ˙OH addition to C5 and C6 sites are kinetically more favorable than to other sites. Moreover, the addition of ˙OH to C5 and C6 sites is highly exothermic with respect to their energies of the reaction complexes, whereas the reactions of other sites are endothermic relevant to their energies of the reaction intermediates. These results imply that the addition of ˙OH to the C5 and C6 sites for 5-caCyt are both thermodynamically and kinetically more favorable than addition to other sites, and have a higher reaction probability. In contrast, the much lower probability is the addition at C2, N3, C4 and C7 sites. These results are in agreement with the experimental results,19 which showed no evidence for addition at C2, N3, C4 and C7 sites. The same conclusion exists in the aqueous phase. Therefore, only the addition of ˙OH to the C5C6 bond for 5-caCyt has been investigated in this paper. Meanwhile, there are five abstractable hydrogen atoms viz. the H3 and H4 attached to N4, the H5 attached to C6, the H6 attached to O4, and the H7 attached to N1. The H7 abstraction reaction here is neglected, and the reason is that N1 of 5-caCyt is always bonded to a carbon in the deoxyribose in the real case of DNA.
3.1 Stationary point structures and energetics in the gas phase
Since ˙OH has extremely high chemical reactivity, various complexes can be formed with 5-caCyt. The two cases in which the H and O of ˙OH are pointed toward the π-face of 5-caCyt have initially been considered, leading to the π-bonded and H-bonded complexes formed in these reactions (see Fig. 2). For the H-bonded complex, the H-bond is formed by the N or O of 5-caCyt and H of ˙OH as well as the H of 5-caCyt and the O of ˙OH. As for a π-bonded complex, the π-bond is formed by the O of ˙OH with C of 5-caCyt. According to the literature,43 the energy of the H-bond depends on the Y⋯H distance and the X–H⋯Y angle (where X is a hydrogen donor and Y is a hydrogen acceptor). According to the Y⋯H distances, all H-bonds can be divided into strong (Y⋯H < 1.600 Å), medium (Y⋯H = 1.600–1.900 Å), and weak (Y⋯H > 1.900 Å) bonds. According to the range of Y⋯H distances and the number of H-bonds, the H-bonds in the complexes should be assigned as weak and medium (as shown in Fig. 2). | ||
| Fig. 2 Optimized structures (bond distances in Å) of the H-bonded and π-bonded complexes for ˙OH-mediated 5-caCyt in the gas phase using the CBS-QB3 composite approach. | ||
In IM1, the distances for the O of ˙OH with C5 and C6 are 2.427 and 2.511 Å, respectively, implying that it is a typical π-bonded complex. Similar to IM1, the distances for the O of ˙OH with C5 and C6 in IM2 are 2.417 and 2.486 Å, respectively, which is also a typical π-bonded complex. Dissimilar with IM1 and IM2, in IM6, the H of ˙OH forms a H-bond with O3 of 5-caCyt at a distance of 1.897 Å, and the O of ˙OH also forms a H-bond with H6 of 5-caCyt at a distance of 1.868 Å, which is a typical H-bonded complex. Identically to IM6, two hydrogen bonds formed in IM5, the distances of C7–O4⋯H1 and H5⋯O1–H1 are 1.987 and 2.279 Å, respectively. For IM3 and IM4, the H-bonded complex formation is mainly aroused from the interactions of the lone pairs on the N3 and O3 atoms with the hydrogen of ˙OH, respectively. According to the relative energy values (ΔEg), the order of stability for the reaction complex is IM6 ≈ IM3 > IM4 > IM2 > IM5 ≈ IM1 (see Table 1).
| System | Species | CBS-QB3b | PCMc | |||
|---|---|---|---|---|---|---|
| ΔEg | ΔGg | ΔGg≠ | ΔGs | ΔGs≠ | ||
| a ΔEg, ΔGg, and ΔGg≠ are the relative energy, relative free energy, and activation free energy in the gas phase, respectively; ΔGs and ΔGs≠ are the relative free energy and activation free energy with the PCM model based on the optimized geometries in the aqueous phase.b CBS-QB3 composite approach.c CBS-QB3 with the PCM model.d Denotes 5-caCyt+˙OH.e Denotes 5-caCytN3+ +˙OH.f Denotes 5-CytCOO− + OH. | ||||||
| Addition reactions | ||||||
| Rd | 0.00 | 0.00 | 0.00 | |||
| IM1 | −7.68 | 23.12 | 27.76 | |||
| TS1 | −11.71 | 25.76 | 28.64 | |||
| P1 | −51.39 | −14.22 | −6.84 | |||
| IM2 | −12.54 | 19.56 | 28.27 | |||
| TS2 | −11.47 | 27.68 | 40.26 | |||
| P2 | −92.61 | −55.84 | −42.00 | |||
| IM1 → P1 | 2.64 | 0.88 | ||||
| IM2 → P2 | 8.12 | 11.99 | ||||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||
| H-atom abstraction reactions | ||||||
| Reactions of ˙OH-mediated 5-caCyt | IM3 | −27.18 | 4.60 | 12.80 | ||
| TS3 | 22.04 | 58.91 | 79.05 | |||
| P3 | −20.80 | 9.70 | −15.23 | |||
| IM4 | −14.29 | 14.35 | 16.22 | |||
| TS4 | 44.49 | 80.46 | 99.23 | |||
| P4 | −28.89 | 1.39 | 1.51 | |||
| IM5 | −8.56 | 20.75 | 27.25 | |||
| TS5 | 24.12 | 112.65 | 66.67 | |||
| P5 | −31.77 | −6.90 | −11.53 | |||
| IM6 | −28.44 | 5.69 | 19.83 | |||
| TS6 | 40.45 | 74.57 | 72.99 | |||
| P6 | −16.48 | 15.21 | −22.36 | |||
| IM3 → P3 | 54.31 | 66.25 | ||||
| IM4 → P4 | 66.11 | 83.01 | ||||
| IM5 → P5 | 91.90 | 39.42 | ||||
| IM6 → P6 | 68.89 | 53.16 | ||||
|
||||||
| Addition reactions | ||||||
| Reactions of ˙OH-mediated 5-caCytN3+ | R′e | 0.00 | 0.00 | 0.00 | ||
| IM1′ | −27.85 | −2.54 | 23.75 | |||
| TS1′ | −4.73 | 32.41 | 41.30 | |||
| P1′ | −53.70 | −15.61 | −2.97 | |||
| IM2′ | −34.81 | −8.46 | 17.03 | |||
| TS2′ | 1.65 | 40.14 | 49.00 | |||
| P2′ | −79.17 | −41.47 | −39.20 | |||
| IM1′ → P1′ | 34.95 | 17.55 | ||||
| IM2′ → P2′ | 48.60 | 31.97 | ||||
|
||||||
| H-atom abstraction reactions | ||||||
| IM3′ | −44.59 | −14.00 | 16.15 | |||
| TS3′ | 38.77 | 74.69 | 103.81 | |||
| P3′ | −47.17 | −23.31 | −2.34 | |||
| IM5′ | −34.81 | −8.45 | 23.35 | |||
| TS5′ | 36.23 | 68.65 | 80.88 | |||
| P5′ | −59.13 | −34.18 | −2.83 | |||
| IM6′ | −31.74 | −4.61 | 17.70 | |||
| TS6′ | 25.01 | 60.12 | 78.98 | |||
| P6′ | 42.17 | −325.88 | 37.74 | |||
| IM3′ → P3′ | 88.69 | 87.66 | ||||
| IM5′ → P5′ | 77.10 | 57.53 | ||||
| IM6′ → P6′ | 64.73 | 61.28 | ||||
|
||||||
| Addition reactions | ||||||
| R′′f | 0.00 | 0.00 | 0.00 | |||
| IM1′′ | −60.31 | −27.01 | 6.35 | |||
| TS1′′ | −35.43 | 5.30 | 18.53 | |||
| P1′′ | −93.21 | −53.11 | −35.78 | |||
| IM2′′ | −60.31 | −27.01 | 6.35 | |||
| TS2′′ | −41.15 | −9.72 | 22.09 | |||
| P2′′ | −116.60 | −78.56 | −59.65 | |||
| IM1′′ → P1′′ | 32.31 | 12.18 | ||||
| IM2′′ → P2′′ | 17.29 | 15.74 | ||||
|
||||||
| H-atom abstraction reactions | ||||||
| Reactions of ˙OH-mediated 5-caCytCOO− | IM3′′ | −53.37 | −18.05 | 4.68 | ||
| TS3′′ | −5.49 | 33.04 | 68.00 | |||
| P3′′ | −86.49 | −55.01 | −30.08 | |||
| IM4′′ | −60.73 | −29.30 | −2.02 | |||
| TS4′′ | 19.61 | 57.76 | 89.24 | |||
| P4′′ | −44.29 | −14.21 | 1.24 | |||
| IM5′′ | −60.31 | −27.01 | 6.35 | |||
| TS5′′ | −31.11 | 3.64 | 38.90 | |||
| P5′′ | −65.24 | −36.44 | −13.68 | |||
| IM3′′ → P3′′ | 51.09 | 63.32 | ||||
| IM4′′ → P4′′ | 87.06 | 91.26 | ||||
| IM5′′ → P5′′ | 30.65 | 32.55 | ||||
Addition reaction mechanism. As mentioned above, the ˙OH can add to C5 and C6 sites of 5-caCyt denoted as R1 and R2, respectively. As seen in Fig. 2 and 3, in path R1, the ˙OH firstly forms a π-bonded complex IM1. With the approaching of ˙OH, the O1⋯C6 bond has been cleaved, and then adds to the C5 site via transition state TS1 with a ΔGg≠ value of only 2.64 kJ mol−1, leading to the formation of P1 (Fig. 4 and Table 1). In P1, the C5 atom has a tetrahedral structure, and the distances of C5–C4, C5–C6, and C5–C7 are stretched to 1.538, 1.477, and 1.536 Å, respectively, longer than those in IM1 (1.462 Å for C5–C4, 1.386 Å for C5–C6, and 1.461 Å for C5–C7), suggesting that the hybridization of the C5 atom converts from sp2 in IM1 to sp3 in P1 and the C5
C6 double bond turns into a single bond. Identically to path R1, the IM2 is converted into P2 via transition state TS2 associated with a ΔGg≠ value of 8.12 kJ mol−1. As seen in Table 1, the ΔEg values of P1 and P2 are −51.39 and −92.61 kJ mol−1, respectively. The spin maxima values also come to the same conclusion that there is higher delocalization of the unpaired electron in P2 (Table 2 and Fig. 5). These indicate that ˙OH addition to the C6 site would be thermodynamically more favorable than to the C5 site. However, there is a small energy barrier for the ˙OH addition to the C6 site, while it is nearly barrierless (2.64 kJ mol−1) for addition to the C5 site. This implies that the ˙OH addition to the C5 site is a little more kinetically favorable than to the C6 site, which agrees with ˙OH addition at the C5 site preferentially in the experiments.19
 | ||
| Fig. 3 Optimized structures (bond distances in Å) in the gas phase for the addition reaction of ˙OH-mediated 5-caCyt (paths R1 and R2) using the CBS-QB3 composite approach. | ||
 | ||
| Fig. 4 The potential energy surfaces (ΔGg in kJ mol−1) along the reaction of ˙OH-mediated 5-caCyt (paths R1–R6) in the gas phase. R denotes 5-caCyt + ˙OH. | ||
| Species | P1 | P2 | P3 | P4 | P5 | P6 |
|---|---|---|---|---|---|---|
| N1 | 0.14 | |||||
| O1 | 0.13 | 0.20 | ||||
| O2 | 0.11 | 0.11 | ||||
| N3 | 0.24 | 0.11 | 0.27 | |||
| C4 | −0.11 | −0.17 | −0.12 | |||
| N4 | 0.80 | 0.72 | ||||
| C5 | 0.72 | 0.13 | ||||
| C6 | 0.64 | 0.86 | ||||
| C7 | ||||||
| O3 | 0.13 | |||||
| O4 | 0.64 |
 | ||
| Fig. 5 The map of spin density distribution for the product radicals of the ˙OH-mediated 5-caCyt in the gas phase. | ||
H-atom abstraction. In this section, the ˙OH is abstracting from the H3 and H4 of the NH2 group, the H5 of the cyclic C6, and the H6 of the COOH group for 5-caCyt, denoted as paths R3–R6, respectively. From Fig. 2, it is already seen that ˙OH can form H-bonded complexes with 5-caCyt and hydrogen abstraction reactions may occur from these complexes. The transition states TS3, TS4, TS5, and TS6 corresponding to the abstraction of H3 in IM3, H4 in IM4, H5 in IM5, and H6 in IM6 are located shown in Fig. 6. In all of the transition states, the abstractable hydrogen atoms are located closer to the ˙OH groups associated with ΔGg≠ values of 54.31, 66.11, 91.90, and 68.89 kJ mol−1, respectively, leading to the formation of product radicals P3, P4, P5, and P6. As seen from Table 1 and Fig. 4, the computed results clearly demonstrate that the H4 and H5 abstractions are slightly favored due to thermodynamic control while the H3 and H6 abstractions can only result from kinetic factors.
 | ||
| Fig. 6 Optimized structures (bond distances in Å) in the gas phase for the H-atom abstraction reaction of ˙OH-mediated 5-caCyt (paths R3–R6) using the CBS-QB3 composite approach. | ||
As seen in Table 2 and Fig. 5, their stabilities are also analyzed by the spin densities. The spin density values of these radical systems are observed mainly on two atoms of 5-caCyt, viz. N3 (0.11) and N4 (0.80) in P3, N3 (0.27) and N4 (0.72) in P4, and C5 (0.13) and O4 (0.64) in P6. It may be noted that except for P5, all systems show the partial spin density transferring from the NH˙ or COO˙ group to the pyrimidine ring, resulting in the distortion of the ring plane or COO˙ group of 5-caCyt under the influence of the unpaired σ electron. While in the case of P5, the spin density is mainly located on C6 (0.86), indicating a relatively localized unpaired σ electron and an unstable structure in theory. However, the P5, based on the ΔGg value, is the most stable (Table 1). As depicted in Fig. 5 and 6, the geometry of P5 remains planar in character in the ring π-system. The NICS(0) value (−5.82) in the center of the six-membered ring for P5 also shows a strong aromaticity involved in the lower energy structure (Table S6†). Although path R5 is a strong exothermic reaction, the ΔGg≠ value is high, suggesting that path R5 in the gas phase is significant to the disadvantage of the abstraction reaction. Thus in the gas phase, it is obvious that path R4 is much more favorable than other abstraction paths and has a high possibility of occurring.
Compared to the addition reaction, the transition states of the abstraction reactions are unstable. The concerned high activation free energies mainly arise from these cyclic transition states. In such cyclic transition states, the orbits required for the bond dissociation and formation are deformed so much that a large amount of deformation energy is substantially needed. Then, it is of great interest whether the activation free energies of the hydrogen abstraction reactions are reduced under strong acid conditions. Similarly, what will ensue from the addition reaction? Then, the reaction of ˙OH mediated 5-caCyt at a lower pH region has been investigated.
The addition of ˙OH to C5 and C6 sites of 5-caCytN3+, are denoted as paths R1′ and R2′, respectively (Fig. 7). With approaching ˙OH toward the C5 and C6 sites of 5-caCytN3+, the complexes IM1′ and IM2′ are formed. Starting from IM1′ and IM2′, the ˙OH are bonded to C5 and C6 sites of 5-caCytN3+ via the transition states TS1′ and TS2′ with the corresponding ΔGg≠ values of 34.95 and 48.60 kJ mol−1, respectively (Fig. 8 and Table 1). Compared to paths R1 and R2, the differences in the structures of the transition states and adduct radicals are very small except for IM1′ and IM2′. As for IM1′, the interaction between the O of ˙OH and the π-face of 5-caCytN3+ results in the formation of a π-bonded complex, whereas for IM2′, the ˙OH moved out from the π-face of 5-caCytN3+ to the molecular plane which eventually leads to the formation of a hydrogen bonded complex, thereby exhibiting that there are obvious differences in the ΔGg≠ values. As seen in Table 1, the increase in the ΔGg≠ value is by 32.31–40.48 kJ mol−1 as compared to those in the paths for 5-caCyt, implying that the paths (R1′ and R2′) are obviously at a disadvantage.
 | ||
| Fig. 7 Optimized structures (bond distances in Å) in the gas phase for the addition reaction of ˙OH-mediated 5-caCytN3+ (paths R1′ and R2′) using the CBS-QB3 composite approach. | ||
 | ||
| Fig. 8 The potential energy surfaces (ΔGg in kJ mol−1) along the reaction of ˙OH-mediated 5-caCytN3+ (paths R1′–R3′, R5′ and R6′) in the gas phase. R′ denotes 5-caCytN3+ + ˙OH. | ||
Identically to the abstraction reaction of 5-caCyt, the ˙OH is closing to the H3, H5, and H6, respectively, and the corresponding H-bonded complexes IM3′, IM5′, and IM6′ are formed (Fig. 9). And then, the H3, H5, and H6 of 5-caCytN3+, respectively, are abstracted via the transition states TS3′, TS5′, and TS6′ with the corresponding ΔGg≠ values of 88.69, 77.10, and 64.73 kJ mol−1, respectively (Fig. 8 and 9). This suggests that path R6′, relative to paths R3′ and R5′, is likely to happen in the gas phase.
 | ||
| Fig. 9 Optimized structures (bond distances in Å) in the gas phase for the H-atom abstraction reaction of ˙OH-mediated 5-caCytN3+ (paths R3′, R5′, and R6′) using the CBS-QB3 composite approach. | ||
As seen in Table 1, it is obvious that the ˙OH addition reactions at a lower pH range have smaller activation free energies, with a range of 16.13–53.74 kJ mol−1, than those of the hydrogen abstraction reaction, suggesting that the reactivity of ˙OH with 5-caCytN3+ is actually dominated by addition. However, the ΔGg≠ values of ˙OH-mediated 5-caCyt paths are higher in the strong acidic condition than in the neutral condition, and then, it is of great concern whether the ΔGg≠ values of these paths will be influenced in alkaline surrounding.
 | ||
| Fig. 10 Optimized structures (bond distances in Å) in the gas phase for the addition reaction of ˙OH-mediated 5-CytCOO− (paths R1′′ and R2′′) using the CBS-QB3 composite approach. | ||
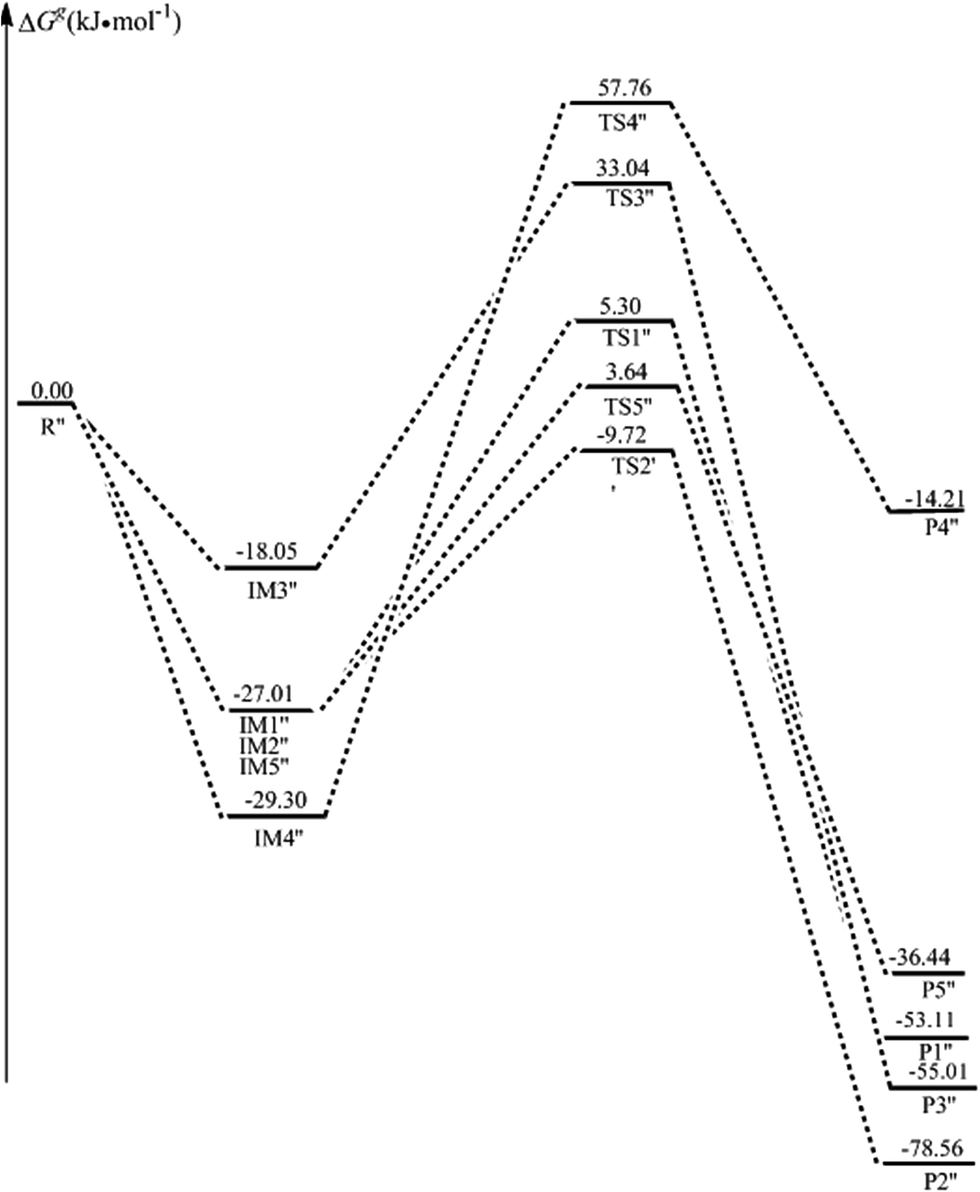 | ||
| Fig. 11 The potential energy surfaces (ΔGg in kJ mol−1) along the reaction of ˙OH-mediated 5-CytCOO− (paths R1′′–R5′′) in the gas phase. R′′ denotes 5-CytCOO− + ˙OH. | ||
 | ||
| Fig. 12 Optimized structures (bond distances in Å) in the gas phase for the H-atom abstraction reaction of ˙OH-mediated 5-CytCOO− (paths R3′′–R5′′) using the CBS-QB3 composite approach. | ||
As mentioned above, the ΔGg≠ values of ˙OH-mediated 5-caCyt are to some degree affected by the acid–base environment. Then, it is of great interest whether the ΔGg≠ values of these paths are further influenced by the contribution of the bulk water.
3.2 Stationary point structures and energetics in the aqueous phase
In modeling solvation effects, an important question arises as to whether computational results are sensitive to the presence of the bulk water. The solvation effect on the ˙OH-mediated 5-caCyt, 5-caCytN3+, and 5-CytCOO−, has been discussed. The important bond lengths of all stationary points for all paths in the aqueous phase are also shown in Fig. S5–S13,† implying that small geometrical changes are induced by the presence of bulk water. The influence of solvation on the activation free energies can be explained by the evolution of the dipole moments for all paths (Table 3).| Reactions of ˙OH-mediated 5-caCyt | |||||||||||
| R1 | μ | R2 | μ | R3 | μ | R4 | μ | R5 | μ | R6 | μ |
| IM1 | 7.32 | IM2 | 4.82 | IM3 | 8.60 | IM4 | 7.17 | IM5 | 5.74 | IM6 | 4.27 |
| TS1 | 6.88 | TS2 | 4.95 | TS3 | 5.46 | TS4 | 7.61 | TS5 | 6.63 | TS6 | 8.28 |
|
|||||||||||
| Reactions of ˙OH-mediated 5-caCytN3+ | |||||||||||
| R1′ | μ | R2′ | μ | R3′ | μ | — | — | R5′ | μ | R6′ | μ |
| IM1′ | 1.11 | IM2′ | 0.79 | IM3′ | 0.99 | — | — | IM5′ | 0.79 | IM6′ | 2.09 |
| TS1′ | 1.56 | TS2′ | 1.29 | TS3′ | 2.86 | — | — | TS5′ | 2.16 | TS6′ | 2.80 |
|
|||||||||||
| Reactions of ˙OH mediated 5-CytCOO− | |||||||||||
| R1′′ | μ | R2′′ | μ | R3′′ | μ | R4′′ | μ | R5′′ | μ | — | — |
| IM1′′ | 2.53 | IM2′′ | 2.53 | IM3′′ | 3.73 | IM4′′ | 4.75 | IM5′′ | 2.53 | — | — |
| TS1′′ | 2.82 | TS2′′ | 2.65 | TS3′′ | 4.73 | TS4′′ | 4.35 | TS5′′ | 2.82 | — | — |
As for the neutral condition, the activation free energy of path R3 in water is 11.94 kJ mol−1 higher than in the gas phase, attributed to the dipole moment of TS3 (μ = 5.46 debye) which is much less than that of IM3 (μ = 8.60 debye), and the transition state TS3 is unstabilized in water by solvation. This indicates that water has a significantly negative catalytic effect on path R3. Dissimilar with path R3, the dipole moments (μ = 6.63 debye for TS5 and 8.28 debye for TS6) are larger than their intermediates (μ = 5.74 debye for IM5 and 4.27 debye for IM6), and then the solvation of water on their transition states are stronger than on the intermediates. This can explain why the paths R5 and R6 are associated with lower activation free energies in the aqueous phase than in the gas phase, illustrating that water has a positive catalytic effect on these paths. For R1 and R2, the dipole moments of TS1 and TS2, relative to IM1 and IM2, have a very small change of about 0.13–0.44 debye, indicating that solvation is comparatively negligible.
Note that water also has a significantly negative catalytic effect on path R4. But unlike R3, the dipole moment (μ = 7.61 debye for TS4) is a little bit larger than its intermediate (μ = 7.17 debye for IM4), suggesting that the solvation of water on its transition state is a little stronger than on the intermediate. By that analogy, the activation free energy of path R4 should be a bit lower in the aqueous phase than in the gas phase. However, the dipole moment approach obviously appears to fail. For this reason, the NPA charges are introduced. The O of ˙OH has strong electronegativity, and the increase in negative charge for the oxygen atom shows a high reactivity toward the hydrogen donor. The present computed NPA charges for O of ˙OH for IM4 in the gas and aqueous phases are −0.415 and 0.287 e, respectively (Table S8†), which may explain why path R4 in the aqueous phase is associated with a poorer reaction trend than in the gas phase.
As for the strong acid condition, the solvation has more or less effect on the ˙OH-mediated 5-caCytN3+. The activation free energies of paths R1′–R3′, R5′ and R6′, are reduced from 34.95, 48.60, 88.69, 77.10, and 64.73 kJ mol−1 to 17.55, 31.97, 87.66, 57.53, and 61.28 kJ mol−1, respectively, illustrating that water has a positive catalytic effect on these paths. The same conclusion is obtained from the evolution of the dipole moments. As for the alkaline condition, the solvation still has to some extent an effect on the ˙OH-mediated 5-CytCOO−. The ΔGs≠ values of paths R1′′–R5′′ are 12.18, 15.74, 63.32, 91.26, and 32.55 kJ mol−1, illustrating that water has a positive catalytic effect on the addition reaction, whereas the solvation effect is reversed for the H5 atom abstraction.
As seen in Table 1, as for the addition, 5-caCyt is nearly barrierless for the C5 channel and has a small barrier of 11.99 kJ mol−1 for the C6 route. Similarly, the observed small difference also exists in the 5-caCytN3+ and 5-CytCOO− paths. These results indicate some amount of regioselectivity, which agrees with the tendencies found experimentally.19 As for the abstraction reaction, the H5 atom is easier to be abstracted in neutral, acid and alkaline conditions. Moreover, the H5 abstraction in alkaline media might be competitive with the addition reactions, having a ΔGs≠ value of 32.55 kJ mol−1, which is only 17–20 kJ mol−1 more energetic than the results for the addition reactions.
4. Summary and conclusions
Three distinct groups of ˙OH-mediated 5-caCyt mechanisms have been explored in this work: the direct reaction (paths R1–R6), the acid-induced process (paths R1′–R3′, R5′ and R6′), and the alkaline-catalytic reaction (paths R1′′–R5′′). Meanwhile, these paths have been further investigated by the contribution of bulk water.(1) The addition of ˙OH to the C5C6 double bond of 5-caCyt is the most favourable in neutral, acid and alkaline conditions, and the ΔGs≠ value of the C5 channel is a little higher than that of the C6 route. The observed small differences in the activation free barriers indicate some amount of regioselectivity,15g–j,44 which also agrees with the tendencies found experimentally.19 This is also in agreement with the conclusions of the ˙OH-mediated cytosine reaction reported experimentally and theoretically.45
(2) The differences in ΔGs≠ values for the ˙OH addition to neutral, deprotonated and N3-protonated 5-caCyt are small, implying that the discrimination is not obviously from an energetic point of view. The ΔGs≠ values of the N3-protonated 5-caCyt system are slightly higher, indicating that the reaction trends are a little weakened.
(3) Concerning the H-atom abstractions, the H5 atom at the C6 position seems to be more labile than abstraction at other positions in neutral, acid and alkaline conditions. Moreover, H5 abstraction in alkaline media might be competitive with the addition reactions, having a ΔGs≠ value of 32.55 kJ mol−1, which is only 17–20 kJ mol−1 more energetic than for the addition reactions. As far as we know, this is the first theoretical report unveiling the effect of acid–base environments in radical reactions, which is also likely to be a new insight for the reactivity of nucleosides with a hydroxyl radical.
5. Final remarks
As seen in Table 1, addition to the C5C6 double bond is more favourable. Concerning the H-atom abstractions, the H atom at the C6 position seems to be more labile than abstraction at other positions according to the present results. Meanwhile, it is clear that addition and H5 atom abstraction for 5-caCyt are prone to happen in neutral or alkaline media, suggesting that DNA bases are easily damaged when exposed to a surrounding of hydroxyl radicals in such case. On the contrary, the reaction trend of ˙OH-mediated 5-caCyt is somewhat weakened at very low pH. Moreover, the product radical formed by 5-caCyt and ˙OH in the aqueous phase is less stable in the acidic condition than in neutral and alkaline conditions, hinting that it is not necessary to decompose these product radicals in harsh conditions. Then DNA bases may be reprieved from oxidation at low pH. The 5-caCyt at the N3 position is easily protonated at pH < 1. However, the DNA macromolecule should be hydrolysed in this strongly acidic pH, even at milder conditions. Therefore, some protective measures for DNA bases should be taken. For example, some antioxidants should be added in mammalian brain tissues. The reason is that many antioxidants can protect biomolecules against DNA damage. However, antioxidant protection against free radicals should be taken with caution since the antioxidant action might actually stimulate cancer progression through the enhanced survival of tumour cells. Of course it would be better to avoid all the radicals for DNA bases. This work might provide some implications for clarifying the reasons for these diseases caused by ˙OH mediated damage to biomolecules.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No.: 21473108), Shaanxi Innovative Team of Key Science and Technology (2013KCT-17), and the Introducing Talents Foundation of Shaanxi University of Technology (No.: SLGKYQD2-13, SLGQD14-10).References
- J. A. Theruvathu, C. T. Aravindakumar, R. Flyunt, J. von Sonntag and C. von Sonntag, J. Am. Chem. Soc., 2001, 123, 9007 CrossRef CAS PubMed.
- Q. Y. Cheng, J. D. Gu, K. R. Compaan and H. F. Schaefer, Chem.–Eur. J., 2010, 16, 11848 CrossRef CAS PubMed.
- T. Finkel and N. J. Holbrook, Nature, 2000, 408, 239 CrossRef CAS PubMed.
- B. N. Ames, M. K. Shigenaga and T. M. Hagen, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 7915 CrossRef CAS.
- V. K. C. Ponnaluri, J. P. Maciejewski and M. Mukherji, Biochem. Biophys. Res. Commun., 2013, 436, 115 CrossRef CAS PubMed.
- H. Zhao and T. Chen, J. Hum. Genet., 2013, 58, 421 CrossRef CAS PubMed.
- L. Shen and Y. Zhang, Curr. Opin. Cell Biol., 2013, 25, 289 CrossRef CAS PubMed.
- S. Schiesser, B. Hackner, T. Pfaffeneder, M. Mǖller, C. Hagemeier, M. Truss and T. Carell, Angew. Chem., Int. Ed., 2012, 51, 6516 CrossRef CAS PubMed.
- C. P. Walsh and G. L. Xu, Curr. Top. Microbiol. Immunol., 2006, 301, 283 CAS.
- C. G. Spruijt, F. Gnerlich, A. H. Smits, T. Pfaffeneder, P. W. T. C. Jansen, C. Bauer, M. Münzel, M. Wagner, M. Müller, F. Khan, H. C. Eberl, A. Mensinga, A. B. Brinkman, K. Lephikov, U. Müller, J. Walter, R. Boelens, H. van Ingen, H. Leonhardt, T. Carell and M. Vermeulen, Cell, 2013, 152, 1146 CrossRef CAS PubMed.
- M. W. Kellinger, C. X. Song, J. Chong, X. Y. Lu, C. He and D. Wang, Nat. Struct. Mol. Biol., 2012, 19, 831 CAS.
- X. W. Xing, Y. L. Liu, M. Vargas, Y. Wang, Y. Q. Feng, X. Zhou and B. F. Yuan, PLoS One, 2013, 8, 8e72993 CrossRef PubMed.
- H. Wu and Y. Zhang, Cell, 2014, 156, 45 CrossRef CAS PubMed.
- M. Valko, M. Izakovic, M. Mazur, C. J. Rhodes and J. Telser, Mol. Cell. Biochem., 2004, 266, 37 CrossRef CAS.
- (a) C. J. Burrows and J. G. Muller, Chem. Rev., 1998, 98, 1109 CrossRef CAS PubMed; (b) J. Cadet, M. Berger, T. Douki and J. L. Ravanat, Rev. Physiol., Biochem. Pharmacol., 1997, 131, 1 CAS; (c) W. K. Pogozelski and T. D. Tullius, Chem. Rev., 1998, 98, 1089 CrossRef CAS PubMed; (d) J. R. Wagner, C. Decarroz, M. Berger and J. Cadet, J. Am. Chem. Soc., 1999, 121, 4101 CrossRef CAS; (e) J. Cadet, T. Douki and J. L. Ravanat, Acc. Chem. Res., 2008, 41, 1075 CrossRef CAS PubMed; (f) G. Pratviel and B. Meunier, Chem.–Eur. J., 2006, 12, 6018 CrossRef CAS PubMed; (g) J. R. Wagner and J. Cadet, Acc. Chem. Res., 2010, 43, 564 CrossRef CAS PubMed; (h) Y. J. Ji and Y. Y. Xia, Int. J. Quantum Chem., 2005, 101, 211 CrossRef CAS PubMed; (i) F. M. Antonio, M. Manuela and R. S. Daniel, J. Phys. Chem. B, 2014, 118, 2932 CrossRef PubMed; (j) A. Adhikary, A. Kumar, A. N. Heizer, B. J. Palmer, V. Pottiboyina, Y. Liang, S. F. Wnuk and M. D. Sevilla, J. Am. Chem. Soc., 2013, 135, 3121 CrossRef CAS PubMed.
- (a) J. Cadet, T. Delatour, T. Douki, D. Gasparutto, J. P. Pouget, J. L. Ravanat and S. Sauvaigo, Mutat. Res., 1999, 424, 9 CrossRef CAS; (b) C. Chatgilialoglu, M. D. Angelantonio, M. Guerra, P. Kaloudis and Q. G. Mulazzani, Angew. Chem., 2009, 121, 2248 CrossRef PubMed.
- S. Zuo, R. J. Boorstein and G. W. Teebor, Nucleic Acids Res., 1995, 23, 3239 CrossRef CAS PubMed.
- C. von Sonntag, The chemical basis for radiation biology, Taylor & Francis, London, 1987 Search PubMed.
- D. K. Hazra and S. Steenken, J. Am. Chem. Soc., 1983, 105, 4380 CrossRef CAS.
- M. Sumino, A. Ohkubo, H. Taguchi, K. Seio and M. Sekine, Bioorg. Med. Chem. Lett., 2008, 18, 274 CrossRef CAS PubMed.
- S. Irrera and G. Portalone, J. Mol. Struct., 2013, 1050, 140 CrossRef CAS PubMed.
- A. Maiti, A. Z. Michelson, C. J. Armwood, J. K. Lee and A. C. Drohat, J. Am. Chem. Soc., 2013, 135, 15813 CrossRef CAS PubMed.
- M. Chen and J. K. Lee, J. Org. Chem., 2014, 79, 11295 CrossRef CAS PubMed.
- Z. A. Shabarova and A. A. Bogdanov, Advanced Organic Chemistry of Nucleic Acids, Wiley, 1994, p.185 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. J. Zheng, L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc, Wallingford, CT, 2009 Search PubMed.
- J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 1999, 110, 2822 CrossRef CAS PubMed.
- A. G. Baboul, L. A. Curtiss, P. C. Redfern and K. Raghavachari, J. Chem. Phys., 1999, 110, 7650 CrossRef CAS PubMed.
- B. Sirjean and R. Fournet, J. Phys. Chem. A, 2012, 116, 6675 CrossRef CAS PubMed.
- B. Sirjean, P. A. Glaude, M. F. Ruiz-Lòpez and R. Fournet, J. Phys. Chem. A, 2006, 110, 12693 CrossRef CAS PubMed.
- B. Sirjean, P. A. Glaude, M. F. Ruiz-Lòpez and R. Fournet, J. Phys. Chem. A, 2009, 113, 6924 CrossRef CAS PubMed.
- E. K. Pokon, M. D. Liptak, S. Feldgus and G. C. Shields, J. Phys. Chem. A, 2001, 105, 10483 CrossRef CAS.
- J. W. Ochterski, G. A. Petersson and J. A. Montgomery Jr, J. Chem. Phys., 1996, 104, 2598 CrossRef CAS PubMed.
- M. R. Nyden and G. A. Petersson, J. Chem. Phys., 1981, 75, 1843 CrossRef CAS PubMed.
- G. A. Petersson, A. Bennett, T. G. Tensfeld, M. A. Al-Laham, W. Shirley and J. Matzaris, J. Chem. Phys., 1988, 89, 2193 CrossRef CAS PubMed.
- G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081 CrossRef CAS PubMed.
- G. A. Petersson, A. K. Yee and A. Bennett, J. Chem. Phys., 1985, 83, 5105 CrossRef CAS PubMed.
- J. A. Montgomery, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 1994, 101, 5900 CrossRef CAS PubMed.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS PubMed.
- E. Cancès, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef PubMed.
- (a) B. Ośmiałowski, E. Kolehmainen and R. Gawinecki, Magn. Reson. Chem., 2001, 39, 334 CrossRef PubMed; (b) M. Szafran, A. Komasa, A. Katrusiak, Z. Dega-Szafran and P. Barczyński, J. Mol. Struct., 2007, 844, 102 CrossRef PubMed; (c) M. Senyel, O. Alver and C. Parlak, Spectrochim. Acta, Part A, 2008, 71, 830 CrossRef PubMed; (d) A. R. Katritzky, N. G. Akhmedov, J. Doskocz, C. D. Hall, R. G. Akhmedova and S. Majumder, Magn. Reson. Chem., 2007, 45, 5 CrossRef CAS PubMed; (e) C. Parlak, O. Alver and M. Senyel, Spectrochim. Acta, Part A, 2008, 69, 1252 CrossRef PubMed; (f) A. R. Katritzky, N. G. Akhmedov, J. Doskocz, P. P. Mohapatra, C. D. Hall and A. Güven, Magn. Reson. Chem., 2007, 45, 532 CrossRef CAS PubMed.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 CrossRef CAS PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- W. W. Cleland and M. M. Kreevoy, Science, 1994, 264, 1887 CAS.
- (a) K. P. Prasanthkumar, C. H. Suresh and C. T. Aravindakumar, Radiat. Phys. Chem., 2012, 81, 267 CrossRef CAS PubMed; (b) S. Naumov and C. von Sonntag, Radiat. Res., 2008, 169, 355 CrossRef CAS PubMed.
- S. V. Jovanovic and M. G. Simic, J. Am. Chem. Soc., 1986, 108, 5968 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The relevant energy information on different protonated 5-caCyt isomers both in the gas and aqueous phases is listed in Table S1. The comparison of the activation free energies by G3B3 and CB3-QB3 composite approaches are listed in Table S2. Spin contamination (〈S2〉) and after spin annihilation (〈Sa2〉) values in ˙OH-mediated 5-caCyt, 5-caCytN3+ and 5-CytCOO− reactions are listed in Table S3. The relevant energy information on different 5-caCyt isomers both in the gas and aqueous phases is listed in Table S4. The relative energies for the addition of ˙OH to C2, N3, C4, C7 sites of 5-caCyt both in the gas and aqueous phases are listed in Table S5. The nucleus-independent chemical shifts (NICS(0)) for the product radicals of ˙OH abstraction from 5-caCyt in the gas phase are given in Table S6. The relative energies for the addition of ˙OH to C2, C4, C7 sites and the H4 atom abstraction of 5-caCytN3+ both in the gas and aqueous phases are listed in Table S7. The NPA charge on O of ˙OH for path R4 in the gas (a) and aqueous phases (b) are listed in Table S8. The corresponding geometrical structures of protonated 5-caCyt and 5-caCyt isomers in the aqueous phase are listed Fig. S1 and S2. The potential energy surface (ΔGg in kJ mol−1) along the addition of ˙OH to C2, N3, C4 and C7 sites of 5-caCyt in the gas phase is listed Fig. S3. The potential energy surface (ΔGg in kJ mol−1) along the addition of ˙OH to C2, C4 and C7 sites and the abstraction H4 atom of 5-caCytN3+ in the gas phase is listed Fig. S4. The important bond lengths of all stationary points for the main addition and hydrogen abstraction paths in neutral, acidic and alkaline conditions in the aqueous phase are listed in Fig. S5–S13. See DOI: 10.1039/c5ra17393k |
| This journal is © The Royal Society of Chemistry 2015 |