
Elaborating ordered silicon carbide nanorods by preceramic polymer nanocasting†
Thibaud Nardin,
Julien Cambedouzou*,
Johann Ravaux,
Cyrielle Rey,
Daniel Meyer and
Olivier Diat
Institut de Chimie Séparative de Marcoule, UMR 5257 CEA/CNRS/UM/ENSCM, BP17171, F-30207 Bagnols-sur-Cèze, France. E-mail: julien.cambedouzou@enscm.fr
First published on 7th October 2015
Abstract
Meso- and micro-porous silicon carbide (SiC) ceramics have been successfully synthesized via a nanocasting process. These materials exhibit very high specific surface areas (from 240 to 760 m2 g−1) and present morphologies of highly ordered SiC nanorods. Liquid allylhydropolycarbosilane or poly-1,3,5-trisilacyclohexane (pTSCH) were used as starting pre-ceramic polymers and were casted in a meso-porous silica template (SBA-15) which has undergone densification treatment. After thermal conversion, silica hard templates were subsequently removed by an aqueous hydrofluoric acid treatment leading to SiC inverse replicas of the template. The prepared meso-porous SiC products were thoroughly analyzed using small and wide angle X-ray scattering, electronic microscopies and nitrogen physisorption experiments. From our observations, the preparation procedure preserves the nanoscale structure of the silica templates and leads to highly porous SiC materials with tunable morphologies and specific surface areas.
Introduction
Silicon carbides (SiC) represent a hot topic of study because of their unique properties such as temperature stability, oxidation resistance or thermal conductivity. They make them attractive for several fields of applications.1–4 SiC are especially considered as catalyst supports,5–7 or for nuclear fuel cladding.8The requirement for high specific surface area (SSA) and tunable morphology in these applications makes the elaboration of meso- and micro-porous SiC an attractive challenge. Furthermore, the demand of well-ordered nanostructured ceramic materials is increasing in a wide range of applications such as membranes for liquid metal or gas filtration, biomedical applications, tissue engineering or interpenetrating composites.9–12
However, the control of their morphology and/or porosity is often difficult as long as carbothermal reduction is considered for their synthesis.13 Meanwhile, the elaboration of SiC from pre-ceramic polymers such as polycarbosilane14–16 offers remarkable possibilities in order to obtain meso-porous materials.
In 2004, Krawiec et al. have proposed a synthesis via chemical vapor infiltration of meso-porous silica (MCM-48 and SBA-15) and obtained disordered meso-porous SiC with high SSA after pyrolysis and silica matrix removal.17 The same year, Park et al. have elaborated nano-porous SiC by using a network of silica nanometric spheres (with a diameter from 20 to 100 nm) as template and an allylhydridopolycarbosilane as SiC precursor.18 Relatively dense SiC were obtained with a distribution of meso- and micro-pores and a specific surface area up to 620 m2 g−1. Today, spherical particles of silica are still used for hard templating as reflected in the work of Hoffmann et al. published in 2012.19 Beyond silica beads, many meso-porous silicas have been used for the nanocasting approach. Thus, in 2008, Krawiec et al. have elaborated inverse replicas of meso-porous silica such as KIT-6 (ref. 20) by nanocasting of polycarbosilane. They obtained porous SiC with a specific surface area close to 1000 m2 g−1.21 During the following years, the nanocasting process of this kind of meso-porous materials was the subject of numerous scientific publications addressing various issues: modification of the preceramic polymer, morphological control or pyrolysis temperature.22–25 Hoffmann et al. also worked on the diversification of the solid templates using for example meso-porous borosilicate glasses instead of silica.26 Finally, the nanocasting of polycarbosilane in solid matrices allows the elaboration of SiC with various morphologies such as fibers, tubes and foams.27–29 However, even though a large diversity of solid templates is available for the nanocasting, the choice of pre-ceramic polymers is restricted. We therefore introduced here a liquid polycarbosilane derived from the dehydrogenative coupling of 1,3,5-trisilacyclohexane (pTSCH, see in ESI, Fig. S1†).30 This polymer was compared to a commonly used polycarbosilane of commercial denomination “SMP10” for SiC elaboration by nanocasting into SBA-15.21,31 Pre-ceramic polymers were infiltrated into the meso-porous silica template and, after thermal conversion and template removal, meso- and micro-porous SiC materials were obtained. The polymer derived-ceramics were then characterized and compared at the micro- and nano-metric scale. We finally demonstrated that meso-porous materials exhibiting very high SSA (240–760 m2 g−1) and featuring ordered SiC nanorods can be produced following our original procedure. The latter protocol differs from the usual methods involving carbothermal reduction32 or growing on an oxide template.33
Experimental
Chemicals
Triblock poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) copolymer Pluronic P123 (EO20PO70EO20) and tetraethyl orthosilicate (TEOS) were purchased from Sigma-Aldrich as precursors for the elaboration of SBA-15 mesoporous silica. SiC molecular precursor 1,3,5-trisilacyclohexane (TSCH) was purchased from Gelest, and SMP10 carbosilane oligomers were purchased from Starfire® Systems. No further purification treatment was applied for these products.Synthesis of hard templates
The mesoporous silica SBA-15 template was prepared by hydrothermal synthesis method according to established procedures.34 Thus, the triblock copolymer P123 was dissolved into a concentrated aqueous solution of hydrochloric acid. Then the silica precursor (TEOS) was added and the fluid was stirred at 40 °C for 20 hours. The mixture was subsequently matured at 100 °C during 48 h. After a water washing and a filtration, the product was pyrolyzed at 550 °C for 5 hours. SBA-15 was obtained as a white micrometric powder.The SBA-15 powder was then dispersed into ethanol and densified by centrifugation at 4500 rpm (75 Hz). After drying, a compact powder is obtained and noted SBA-15(c). SBA-15 was also used as a solid template by shaping the SBA-15 powder into pellets under uniaxial compression at 0.2 or 1 GPa. The resulting materials are respectively named SBA-15(2t) and SBA-15(10t). Prior to be used as templating structures, these three different porous materials were dried at 100 °C under vacuum.
SiC synthesis
SiC materials were prepared from the TSCH or SMP10 precursors. In order to obtain the preceramic polymer, TSCH was polymerized at room temperature by a catalytic dehydrogenative coupling in a Schlenk flask under argon atmosphere using bis(cyclopentadienyl)-bis(diphenoxy)titanium as catalyst prefunction.35,36 This catalyst was synthesized using an adapted protocol proposed by Andrä.37 The dehydrogenative polymerization of TSCH results in the poly-1,3,5-trisilacyclohexane (pTSCH), which can be obtained with various degrees of polymerization depending on the reaction time.27 In the following, the acronym pTSCH corresponds to the product resulting from TSCH polymerization for one hour at 65 °C. Therefore, two polymeric precursors (pTSCH and SMP10) were used in this study for the nanocasting.First, the SBA-15(c, 2t and 10t) templates were infiltrated by an excess of the polymer (SMP10 or pTSCH) by capillarity during a couple of days under inert atmosphere. A heat treatment at 150 °C during 1 hour was used to promote the cross-linking of the chosen pre-ceramic polymer. The pyrolysis of the as-obtained composite was conducted in alumina crucibles under a flow of argon in a tubular furnace. The heating process was programmed for a 100 °C h−1 ramp to 1000 °C followed by a 2 hours plateau at this temperature. SiO2/SiC composites were obtained. The silica matrix was further dissolved in an excess of an ammonium fluoride/hydrofluoric acid mixture (NH4F/HF) under strong stirring, according to the following reaction:
| SiO2(s) + 6HF(aq) → H2[SiF6](aq) + 2H2O(l) |
After 6 hours under strong stirring, the material was washed with by ultra-pure water several times. This silica dissolution process was repeated up to 3 times to ensure complete removal and thus obtain porous SiC. Note that SiC obtained from the SMP10 and pTSCH are respectively denoted SiC-S and SiC-T in the following.
Characterization methods
SiC samples were examined by environmental scanning electron microscopy (SEM) using a FEI Quanta 400 FEG. Nitrogen physisorption measurements were performed using a ASAP 2020 at 77 K, after outgassing at 623 K during 8 hours, reaching a pressure below 1 mm Hg, and specific surface areas were calculated using the BET method. X-ray diffraction (XRD) measurements were performed using a Bruker diffractometer (Model D8 Advance, Cu Kα radiation; λ = 0.154 nm). Small and wide angle X-ray scattering (SWAXS) experiments were carried out on a Xenocs bench using a molybdenum source and delivering a 0.8 × 0.8 mm2 monochromatic and collimated beam of 0.071 nm wavelength. Measurements were performed in transmission geometry and the scattering patterns were recorded using an online two-dimensional imaging plate detector (MAR-345) located at a distance of 750 mm from the sample position. A standard procedure for the data treatment was applied, involving an azimuthal averaging and background and empty cell subtraction taking into account the normalization by the acquisition time and the sample transmission. Thermogravimetric analyses (TGA) were performed in a Setsys 1750 CS Evol under argon atmosphere and using a heating rate of 5 °C min−1. Transmission Electron Microscopy (TEM) was performed on a JEOL 2200 FS operating at 200 kV. Samples obtained after heat treatment were ground into a powder and dispersed in ethanol. Samples were deposited on 400-mesh carbon-coated copper grids. After ethanol evaporation, the grid was submitted to analysis.Results and discussion
Bulky SiC and SBA-15 characterization
The thermal conversion at 1000 °C of both pTSCH and SMP10 leads to a SiC of low crystallinity. XRD patterns indeed exhibit broad Bragg peaks corresponding to the crystalline structure of a face-centered cubic β-SiC38 with a lattice constant of ∼0.43 nm (see in ESI, Fig. S2†).Nitrogen physisorption analysis allowed us to determine the SSA of the SBA-15 material, giving a value of 545 m2 g−1. TEM images presented in Fig. 1 show that the sample is under the form of a micrometric powder with grains having hexagonal-base morphology. The mean grain length is ∼1 μm and the mean grain width is ∼0.3 μm. The internal structure of the grain is a 2D hexagonal network of mesoscopic cylindrical pores. These meso-pores are known to be partially interconnected by micro-pores.39
 | ||
| Fig. 1 TEM images of SBA-15. | ||
The meso-pore diameter determined by nitrogen physisorption is about 6.3 ± 1 nm and the 2D-hexagonal lattice parameter is about 11.1 nm, as shown below by SWAXS results. The densified SBA-15 resulting from the centrifugation or uniaxial compression up to 1 GPa possesses the same SSA and the same 2D hexagonal structure of nanometric pores.
Mesoporous SiC
The nanocasting process described above and schematized in Fig. 2 is applied to both polymers using the three solid templates resulting from the SBA-15 densification, namely SBA-15(c), SBA-15(2t) and SBA-15(10t).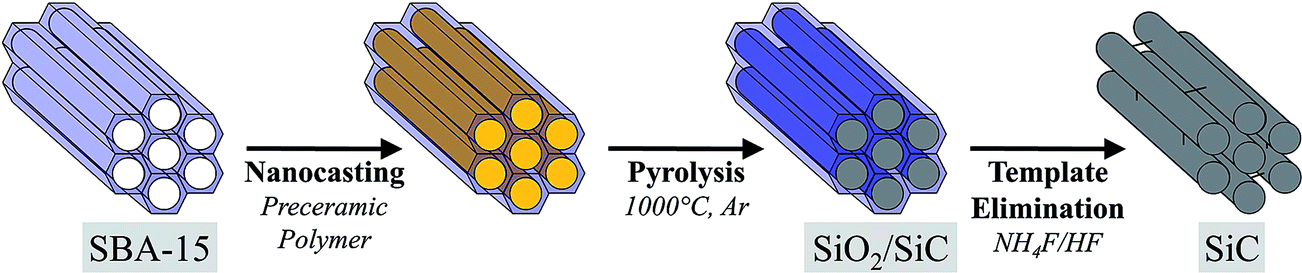 | ||
| Fig. 2 Schematic illustration of the SiC synthesis by nanocasting into SBA-15. | ||
Using either pTSCH or SMP10 as SiC precursors, six final materials are obtained and will be referred to according to the nomenclature given in Table 1. The first macroscopic observation is that the SiC materials resulting from the SMP10 nanocasting are monolithic materials while the ones obtained with pTSCH nanocasting led to SiC powders. In both cases, the SSA of the final material seems to be dependent on the densification method, with a higher SSA for higher applied pressure during the densification. Moreover, for a similar densification process, nanocasting with pTSCH leads to materials with higher SSA than with SMP10. One can also observe that the pTSCH nanocasting process using the highly densified SBA-15(10t) template presents a very low yield after silica dissolution. This observation can potentially be interpreted by a relatively low wettability of the template SBA-15(10t) with regards to the pTSCH precursor.
| Final SiC materials | Pre-ceramic polymer | Solid template | Specific surface area (m2 g−1) | General morphology |
|---|---|---|---|---|
| SiC-Sc | SMP10 | SBA-15(c) | 240 | Monoliths |
| SiC-S2t | SBA-15(2t) | 634 | SSA dependent on the densification method | |
| SiC-S10t | SBA-15(10t) | 751 | ||
| SiC-Tc | pTSCH | SBA-15(c) | 402 | Micrometric powders |
| SiC-T2t | SBA-15(2t) | 763 | ||
| SiC-T10t | SBA-15(10t) | n.a. |
Depending on the template nature, SiC-S exhibits a SSA from 240 to 751 m2 g−1 and SiC-T from 402 to 763 m2 g−1 as shown in Table 1. Although these SSA present the same order of magnitude, the electronic microscopic study of the nanocasted material at the micrometric scale reveals various morphologies and nano-structurations (Fig. 3). SiC-S series exhibit alveolar morphologies and SiC-T a granular morphologies with a grain size similar to that of the SBA-15. Provided that the template is a powder constituted of micrometric grains, we think that the size of the inter-grains spacing has a significant impact on the final morphology of the SiC material. Concerning the materials resulting from SMP10 conversion, the SMP10 precursor will fill all the accessible inter-grains free spaces to form alveolar SiC where each alveolus is a replica of one SBA-15 grain. The resulting material can therefore be viewed as alveoli surrounded by domains of dense SiC, which correspond to the initial inter-grains spaces. SiC nanorods are observed inside these alveoli and result from the inverse replica of the cylindrical meso-pores of SBA-15. Concerning the materials resulting from the pTSCH conversion, the granular morphology of the SiC-S suggests that the inter-grains spacing is not replicated by the pTSCH. Thus the relatively high SSA of SiC-S series suppose that these grains are meso- and/or micro-porous. Unfortunately, SEM images do not allow us to determine whether the hexagonal network of meso-pores has been replicated or not. The SiC nanorods network is only revealed in TEM images (see in ESI, Fig. S6 and S7†).
 | ||
| Fig. 3 SEM images of (a) SiC-Sc, (b) SiC-S10t and (c) SiC-T2t. | ||
To get further insight into the structural organization at the mesoscopic scale, SWAXS measurements were carried out and the corresponding results are shown in Fig. 4. Each diagram was measured after a step of the nanocasting process. The chemical structure is followed during the process by looking at the evolution of the wide angles part of the SWAXS diagram (from q ∼ 6 up to 30 nm−1). The large peaks at q ∼ 11.6, 16.1 and 25.0 nm−1 correspond to the signature of the different molecular or crystalline structures in presence, namely the pre-ceramic polymers, SiO2, and β-SiC (through its (111) reflection), respectively. We can clearly see for SiC-S10t and SiC-T2t materials that the pre-ceramic polymeric scattering feature at 11.6 nm−1 is present after impregnation and disappears after the thermal treatment. Similarly, the SiO2 contribution disappears after the template dissolution with the acidic mixture. Finally, the final structure at the molecular scale is characterized by the β-SiC (111) peak, which is expected for this type of conversion. In the small scattering angle region of the diagram (up to q ∼ 6 nm−1), the 2D hexagonal order of the cylindrical pores in SBA-15 is observed. The corresponding Bragg peaks are indexed in the left part of Fig. 4. This long-range spatial correlation is conserved through the whole process. The decreasing of relative intensity of the peaks between the SBA-15 and the final SiC is due to several reasons. The first decrease of Bragg peak intensity results from the filling of the SBA-15 pores with pre-ceramic polymers, which reduces the contrast between the pores and the walls. The thermal treatment does not strongly affect the peaks intensity, but the final acidic treatment in order to remove the silica template causes a slight intensity decrease. The latter effect might be due to a partial destruction of mesostructured SiC. Note however that due to the granular texture of the samples, the analysis of the SAXS intensities can be only qualitative. Concerning Bragg peak positions, we observe a slight shift to larger scattering angles after the thermal treatment, mainly due to the contraction of the entire solid structure (SiO2 and SiC)40,41 of about 10%. Then, after the SiO2 matrix dissolution, this shift is still observable.
 | ||
| Fig. 4 SWAXS curves through the nanocasting process leading to the final materials SiC-S10t (left) and SiC-T2t (right). The grey dot curve corresponds to the SBA-15, the black curve (i) to the SBA-15 impregnated by the pre-ceramic polymer, the red curve (ii) to the composite SBA-15/SiC after thermal conversion of the latter material, and the blue curve (iii) to the final SiC material. | ||
The nitrogen physisorption isotherms of the final SiC materials are given in Fig. 5. According to the IUPAC classification,42,43 the SBA-15 nitrogen physisorption curve exhibits a type IV isotherm with a H1 hysteresis loop, which highlights a meso-porous material with a narrow distribution of pore sizes. SiC-Sc presents an isotherm with approximately the same appearance than SBA-15, but with a H1 hysteresis shifted to higher pressures. SiC-Tc presents a type I isotherm and a small hysteresis at high pressure. Such an isotherm is characteristic of a micro-porous material. Both isotherms exhibit a narrow hysteresis corresponding to a narrow distribution of meso-pores. SiC replica resulting from the hard templating of SBA-15(2t) and SBA-15(10t) exhibit type [I + IV] isotherms with wide hysteresis loops. Thereby SiC-T2t, SiC-S2t and SiC-S10t are micro- and meso-porous materials with a broad distribution of meso-pore sizes.
 | ||
| Fig. 5 Nitrogen physisorption isotherms of the final materials. | ||
Using the t-plot method of de Boer,44–46 the ratio of SSA assigned to the meso-porosity (SSAmeso) can be calculated and the SSAmicro assigned to the micro-porosity can be deduced from the relation:
| SSA = SSAmeso + SSAmicro |
These results are given in the Table 2.
| Materials | Isotherm type | SSA (±10 m2 g−1) | SSAmeso (m2 g−1) | SSAmeso (% of SSA) |
|---|---|---|---|---|
| SBA-15 | IV | 545 | 380 | 70 |
| SiC-Sc | IV | 240 | 161 | 67 |
| SiC-S2t | [I + IV] | 634 | 196 | 31 |
| SiC-S10t | [I + IV] | 751 | 188 | 25 |
| SiC-Tc | I | 402 | n.a. | n.a. |
| SiC-T2t | [I + IV] | 763 | 184 | 24 |
Describing the pore morphology in the as produced SiC materials is slightly more complicated than for the SBA-15 matrices. As a matter of facts, SBA-15 mesopores are cylinders, and the micropores can roughly be approximated by smaller cylinders (Fig. 6). In the case of SiC, the porosity is much more interconnected and it corresponds to the empty voids between SiC nanorods. In such geometry, discriminating meso-pores and micro-pores becomes difficult. Therefore, it is complex to interpret the value of the Table 2. The first observation is that the SSAmeso of the final SiC are always smaller (by a factor of 2) than those of the SBA-15 templates and the SiC inverse replicas are mainly micro-porous (expect for the SiC-Sc). This result tends to indicate that the peculiar morphology of bundled SiC nanorods, mainly representative of SiC-S2t, SiC-S10t and SiC-T2t, features micropores rather than mesopores. In the SiC-S series, the more compact the template is, the more the SSA of the inverse replica is high. This is consistent with the increasing of the volume ratio of SiC nanorods with regards to dense SiC.
 | ||
| Fig. 6 Porous networks illustration of the SBA-15 and the final SiC materials. The grey and black regions represent respectively the SiO2 and the SiC. The porous network appears in white with two different pore sizes P1 and P2. | ||
Starting from a silica template with a SSA of 545 m2 g−1, SiC replica were obtained with a SSA up to 763 m2 g−1. This increase of SSA can be easily explained by simple geometric arguments (see in ESI, Fig. S8†). As a matter of facts, the hexagonal structure of SBA-15 features a volume content of silica approximately equal to 71%. Consequently, if we neglect the shrinkage during the thermal conversion, the SiC volume content in the replica of this hexagonal lattice is expected to be of 29%. Moreover, the density of the polymer-derived SiC at 1000 °C is similar to the silica density.41 Since the interface does not change during the nanocasting, the SSA increasing can be simply explained by the decreasing of the solid volume fraction in the SiC material.
Conclusions
We proposed here a synthesis by nanocasting of meso- and micro-porous SiC materials exhibiting ordered nanorods. SBA-15 with different densification treatments have been used as solid silica template. Liquid SMP10 and pTSCH were used as pre-ceramic polymers. After thermal conversion, silica hard templates were subsequently removed by aqueous hydrofluoric acid solution.Two distinct pre-ceramic polymers led to different final materials using the same nanocasting process with SBA-15. The final SiC monoliths resulting from the SMP10 nanocasting possess alveolar morphologies with a SSA ranging from 240 to 751 m2 g−1. These materials are composed of dense SiC regions standing alongside regions of rod-like SiC in 2D hexagonal configuration, conserving the nanoscale structure of the template. The densification method on the starting SBA-15 allows us to modify the volume ratio between these two regions. When using the pTSCH nanocasting, a micrometric powder is obtained with a SSA of 763 m2 g−1. In this case, the morphology as well as the SSA is not tunable but the nanocasting process leads to a final SiC very similar to the starting SBA-15.
Finally, the presented nanocasting approach allows elaborating different SiC compounds for which the porosity and morphology can be tuned by the pre-treatment of the silica mould and the choice of the pre-ceramic polymer.
Acknowledgements
The authors would like to thank the French CNRS NEEDS-Matériaux program (2012-2014) and the French ANR program (FANTA-SiC project, ANR-12-JS08-0010) for financial support on this work. The authors acknowledge B. Corso for his help on X-ray scattering and X. le Goff for his help on TEM measurements at the Service Commun de Microscopie Electronique et Analytique of the Université de Montpellier. P. Bauduin and X. Deschanels are thanked for scientific discussions.Notes and references
- J. B. Casady and R. W. Johnson, Solid-State Electron., 1996, 39, 1409–1422 CrossRef.
- M. Singh, J. Mater. Sci. Lett., 1998, 17, 459–461 CrossRef CAS.
- M. Willander, M. Friesel, Q. U. Wahab and B. Straumal, J. Mater. Sci.: Mater. Electron., 2006, 17, 1–25 CrossRef CAS.
- K. M. Pitman, A. M. Hofmeister, A. B. Corman and A. K. Speck, Astron. Astrophys., 2008, 483, 661–672 CrossRef.
- M. J. Ledoux and C. Pham-Huu, CATTECH, 2001, 5, 226–246 CrossRef CAS.
- R. Moene, M. Makkee and J. A. Moulijn, Appl. Catal., A, 1998, 167, 321–330 CrossRef CAS.
- M. Vannice, Y. Chao and R. Friedman, Appl. Catal., 1986, 20, 91–107 CrossRef CAS.
- S. Bragg-Sitton, K. Barrett, I. van Rooyen, D. Hurley and M. Khafizov, Nucl. Eng. Int., 2013, 58, 37–40 CAS.
- A. J. Rosenbloom, D. M. Sipe, Y. Shishkin, Y. Ke, R. P. Devaty and W. J. Choyke, Biomed. Microdevices, 2004, 6, 261–267 CrossRef CAS.
- R. Yakimova, R. M. Petoral, G. R. Yazdi, C. Vahlberg, A. L. Spetz and K. Uvdal, J. Phys. D: Appl. Phys., 2007, 40, 6435 CrossRef CAS.
- A. A. Gokhale, N. V. R. Kumar, B. Sudhakar, S. N. Sahu, H. Basumatary and S. Dhara, Def. Sci. J., 2011, 61, 567–575 CrossRef CAS.
- Cellular Ceramics, ed. M. Scheffler and I. P. Colombo, Wiley-VCH Verlag GmbH & Co. KGaA, 2005, pp. I–XXV Search PubMed.
- J. A. Lely, US Pat., 2854364 A, 1958.
- R. Corriu, M. Enders, S. Huille and J. Moreau, Chem. Mater., 1994, 6, 15–17 CrossRef CAS.
- R. West, L. David, P. Djurovich, H. Yu and R. Sinclair, Am. Ceram. Soc. Bull., 1983, 62, 899–903 CAS.
- S. Yajima, Y. Hasegawa, J. Hayashi and M. Iimura, J. Mater. Sci., 1978, 13, 2569–2576 CrossRef CAS.
- P. Krawiec, C. Weidenthaler and S. Kaskel, Chem. Mater., 2004, 16, 2869–2880 CrossRef CAS.
- K.-H. Park, I.-K. Sung and D.-P. Kim, J. Mater. Chem., 2004, 14, 3436–3439 RSC.
- C. Hoffmann, T. Biemelt, A. Seifert, K. Pinkert, T. Gemming, S. Spange and S. Kaskel, J. Mater. Chem., 2012, 22, 24841–24847 RSC.
- Y. Sakamoto, T.-W. Kim, R. Ryoo and O. Terasaki, Angew. Chem., Int. Ed., 2004, 43, 5231–5234 CrossRef CAS PubMed.
- P. Krawiec, C. Schrage, E. Kockrick and S. Kaskel, Chem. Mater., 2008, 20, 5421–5433 CrossRef CAS.
- Y. F. Shi, Y. Meng, D. H. Chen, S. J. Cheng, P. Chen, H. F. Yang, Y. Wan and D. Y. Zhao, Adv. Funct. Mater., 2006, 16, 561–567 CrossRef CAS PubMed.
- Z. Ji, W. Han, L. Ye, Y. Jiang, H. Li and T. Zhao, Mater. Lett., 2011, 65, 185–187 CrossRef CAS PubMed.
- S. T. Selvan, S. S. Aldeyab, S. M. J. Zaidi, D. Arivuoli, K. Ariga, T. Mori and A. Vinu, J. Nanosci. Nanotechnol., 2011, 11, 6823–6829 CrossRef CAS PubMed.
- X. Yuan, J. Lü, X. Yan, L. Hu and Q. Xue, Microporous Mesoporous Mater., 2011, 142, 754–758 CrossRef CAS PubMed.
- C. Hoffmann, B. Reinhardt, D. Enke and S. Kaskel, Microporous Mesoporous Mater., 2014, 184, 1–6 CrossRef CAS PubMed.
- J. Garcia, PhD Thesis, Université Montpellier 2, 2008.
- H. Wang, X.-D. Li, T.-S. Kim and D.-P. Kim, Appl. Phys. Lett., 2005, 86, 173104 CrossRef PubMed.
- H. Wang, J.-S. Yu, X. Li and D. Kim, Chem. Commun., 2004, 2352–2353 RSC.
- T. Nardin, B. Gouze, J. Cambedouzou, P. Bauduin, M. W. C. Man, X. Deschanels, D. Bourgeois, D. Meyer and O. Diat, J. Mater. Chem. A, 2015, 3, 3082–3090 CAS.
- L. Borchardt, C. Hoffmann, M. Oschatz, L. Mammitzsch, U. Petasch, M. Herrmann and S. Kaskel, Chem. Soc. Rev., 2012, 41, 5053–5067 RSC.
- C. C. Tang, S. S. Fan, H. Y. Dang, J. H. Zhao, C. Zhang, P. Li and Q. Gu, J. Cryst. Growth, 2000, 210, 595–599 CrossRef CAS.
- X. T. Zhou, H. L. Lai, H. Y. Peng, F. C. K. Au, L. S. Liao, N. Wang, I. Bello, C. S. Lee and S. T. Lee, Chem. Phys. Lett., 2000, 318, 58–62 CrossRef CAS.
- D. Y. Zhao, J. L. Feng, Q. S. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- S. Bourg, R. J. P. Corriu, M. Enders and J. J. E. Moreau, Organometallics, 1995, 14, 564–566 CrossRef CAS.
- J. Garcia, D. J. M. Meyer, D. Guillaneux, J. J. E. Moreau and M. Wong Chi Man, J. Organomet. Chem., 2009, 694, 2427–2433 CrossRef CAS PubMed.
- K. Andrä, J. Organomet. Chem., 1968, 11, 567–570 CrossRef.
- E. Whitney, Nature, 1963, 199, 278–280 CrossRef CAS PubMed.
- A. Galarneau, N. Cambon, F. Di Renzo, R. Ryoo, M. Choi and F. Fajula, New J. Chem., 2003, 27, 73–79 RSC.
- Y. Usami, T. Hongo and A. Yamazaki, J. Porous Mater., 2011, 19, 897–902 CrossRef.
- P. Colombo, G. Mera, R. Riedel and G. D. Sorarù, J. Am. Ceram. Soc., 2010, 93, 1805–1837 CAS.
- S. Brunauer, L. S. Deming, W. E. Deming and E. Teller, J. Am. Chem. Soc., 1940, 62, 1723–1732 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- B. C. Lippens and J. H. de Boer, J. Catal., 1965, 4, 319–323 CrossRef CAS.
- H. Kral, J. Rouquerol, K. S. W. Sing and K. K. Unger, Characterization of Porous Solids, Elsevier, 1988 Search PubMed.
- P. Voogd, J. J. F. Scholten and H. van Bekkum, Colloids Surf., 1991, 55, 163–171 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra17376k |
| This journal is © The Royal Society of Chemistry 2015 |