DOI:
10.1039/C5RA17366C
(Paper)
RSC Adv., 2015,
5, 90193-90201
The effect of zinc oxide (ZnO) addition on the physical and morphological properties of cellulose aerogel beads†
Received
27th August 2015
, Accepted 15th October 2015
First published on 15th October 2015
Abstract
Microsized open porous beads of cellulose were made using the dissolution medium containing mixtures of 7 wt% NaOH and 12 wt% urea and additionally various concentrations of ZnO to study its effect on physical and morphological properties of the cellulose beads formed. It has been observed that such cellulose aerogel beads prepared with lower concentrations of ZnO show shrinkage while drying whereas beads prepared with higher concentrations of ZnO do not exhibit much shrinkage. The dried cellulose aerogel beads were spherical with diameters between 2 and 2.5 mm. The skeletal density of all dried cellulose beads was measured as 1.5 g cm−3. FT-IR spectra reveal that the structure of cellulose I transformed to cellulose II during dissolution and regeneration in a coagulation medium, which was also confirmed from XRD measurements. The beads prepared with a NaOH/urea/ZnO aqueous solution exhibit better thermal stability. We found that the addition of 0.5 wt% ZnO to the NaOH/urea mixture greatly increased the specific surface area of the cellulose beads up to 407 m2 g−1 compared to control cellulose beads (341 m2 g−1). SEM images indicate that a dense nano-fibrillar network structure was formed in the interior of the cellulose aerogel beads prepared with 0.5 wt% ZnO.
1. Introduction
Cellulose is one of the most important, abundant and sustainable polymers available on earth. The development of cellulose based materials received much interest over the last decade due to its biocompatibility, biodegradability and environmentally sustainable nature.1,2 Cellulose has some interesting properties like hydrophilicity, high mechanical strength, good optical appearance, high sorption capacity, relative thermostabilization, and good thermal and chemical stability.3 Due to these excellent properties, cellulose can be shaped into different forms like fibers, films, membranes, nanocomposites, sponges, aerogels and spherical particles or beads. Cellulose based materials can be applied in several fields like drug delivery, separation, water treatment, membrane, package, biomaterials, thickener, barrier film and others.3
Among various forms of cellulose based materials, microbeads received quite considerable interest in the recent past. In general, cellulose beads (CB) are spherical shaped particles within the size range of micro- to millimeter scale.4 Such cellulose beads exhibit high surface area, large porosity and several interesting functional properties due to the presence of large number of reactive hydroxyl functional groups.5 Cellulose beads are widely used in separation media, filtration, purification, column chromatography, carrier medium, enzyme immobilization, etc.6–8 The formation of cellulose bead involves three-step process. In the first step, cellulose is dissolved completely in a suitable solvent and then shaping them as spherical beads by a dropping or dispersion technique followed by regeneration in a coagulation bath having an anti-solvent.9 Among these three steps, the dissolution of cellulose in a suitable solvent requires much attention because of the poor solubility of cellulose in water and most of the organic solvents due to its strong inter- and intra-molecular hydrogen bonding.
The most commonly used solvents for the dissolution of cellulose are DMA/LiCl, N-methylmorpholine-N-oxide, DMSO/ammonium fluorides, 1-butyl-3-methylimidazolium cation based ionic liquids and melts of salt hydrates [Ca(SCN)2·4H2O].10–15 On the other hand, all these solvents have several disadvantages like environmental hazardous, high toxicity, volatility, or are highly expensive. In the recent past, the solubility of cellulose concentration around 8–10 wt% was achieved by utilizing environmental friendly NaOH/water mixtures at a lower temperature of around −10 °C.16 Even though, this solvent system is limited to low molecular weight cellulose molecules. To improve the solubility power of NaOH/water system, some additives such as urea, thiourea can be added, which improves the dissolution power of aqueous NaOH solution.17 The addition of urea and utilization of low temperature improves cellulose dissolution because of the formation of large inclusion complexes (formation from cellulose, NaOH, urea and H2O clusters).18 NaOH/water based system in presence and absence of additives received more interest recently due to its cheap and environment friendly reasons. Furthermore, the addition of small amount of zinc oxide into 7 wt% NaOH and 12 wt% urea system was attempted to improve the cellulose dissolution by Yang et al.19 ZnO is one of the compound exhibiting amphoteric nature, which present as Zn(OH)42− species in strong alkaline solutions. This Zn(OH)42− species are able to form stronger hydrogen bonds with cellulose than that of hydrated NaOH system, which leads to the improvement of cellulose dissolution.20,21 Further, the addition of ZnO not only improves the dissolution of cellulose but also is reported to act as a stabilizer against the gelation of cellulose.22 Navard and co-workers23 argue that the stability of cellulose–NaOH–water solution is not reasonably good and cellulose chains try to re-aggregate rapidly and gelation occurs. According to their investigation, the addition of ZnO to NaOH/water solution results in the formation of smaller particles and the surface of particle are hydrolyzed and a layer of hydroxide (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Zn–OH) is resulted. This hydroxide species shall easily attract water and reduces the number of water molecules near the cellulose chains, which restricts their aggregation and delays the gelation.23,24 It has been observed by Xiong and Duan25 that the addition of little amounts of ZnO (0.5 wt%) to NaOH/urea solution can really improve the dissolution of cellulose. Further, the addition of too much ZnO would decrease the dissolution power of the system. This may be due to the overload of ZnO, which consumes the extra NaOH present in the solvent system. Currently the role of ZnO in the mechanism of cellulose dissolution is still under debate.
Zn–OH) is resulted. This hydroxide species shall easily attract water and reduces the number of water molecules near the cellulose chains, which restricts their aggregation and delays the gelation.23,24 It has been observed by Xiong and Duan25 that the addition of little amounts of ZnO (0.5 wt%) to NaOH/urea solution can really improve the dissolution of cellulose. Further, the addition of too much ZnO would decrease the dissolution power of the system. This may be due to the overload of ZnO, which consumes the extra NaOH present in the solvent system. Currently the role of ZnO in the mechanism of cellulose dissolution is still under debate.
In most cases, cellulose films were developed using NaOH/urea/ZnO recipe. The as prepared films show good optical transmittance, thermal stability and tensile strength compared to films prepared without adding ZnO.20 Most recently, cellulose based ZnO nancomposites were developed by mixing 7 wt% NaOH and 0–1.6 wt% ZnO in aqueous solution. These films exhibit better mechanical, thermal properties and also possess good UV blocking ability and antibacterial activity.26,27 In the present investigation, we tried to develop cellulose aerogel microbeads by utilizing the NaOH/urea/ZnO mixtures in aqueous solution. To the best of our knowledge, there is no report available for the preparation of cellulose aerogel microbeads using NaOH/urea mixtures with the addition of zinc oxide in aqueous solution. The influence and effect of the ZnO concentration on the physical and morphological properties of the as formed cellulose aerogel beads were extensively studied using FT-IR, TGA, N2 adsorption/desorption isotherm and SEM techniques.
2. Experimental section
2.1. Materials
Cellulose powder (medium fiber) was purchased from Sigma-Aldrich, Germany. The degree of polymerization for the cellulose powder was found to be 200.28 Other reagents like zinc oxide (particle size in the range of 75–320 nm, see ESI Fig. S1 and S2†), sodium hydroxide, urea and HCl (37%) were obtained from Merck, Germany. Ethanol (99.5%) was used for solvent exchange before super critical drying. Deionized water was used throughout the experiments. All reagents were used as received.
2.2. Preparation of cellulose aerogel microbeads
The cellulose aerogel microbeads were prepared as follows:20,29 cellulose dissolution medium was prepared by mixing 7 wt% NaOH, 12 wt% urea and various concentration of ZnO in 81 wt% aqueous solution at room temperature and the solution was pre-cooled down to −10 °C. Then, 5 wt% of cellulose powder was added to the pre-cooled solution at room temperature and continuously stirred in an ice bath until it became a homogeneous clear solution. The prepared homogenous cellulose solution was kept at low temperature (<0 °C) until further use to avoid aging. The cellulose beads were formed by dropping cellulose solution into a 2 M HCl coagulation bath through a fine glass dropper with a tip diameter of ∼2 mm at room temperature under magnetic stirring (200 rpm). The distance between the surface of coagulation bath and tip of the dropper was maintained at 2 cm to get uniform sized spherical beads. The completion of coagulation (i.e., formation of clear white cellulose beads) requires minimum 2 to 5 min time duration. The as prepared beads were left in the coagulation bath (HCl) for overnight to complete the regeneration of cellulose. Afterwards, cellulose beads were washed several times with water until a neutral pH was achieved. The beads were completely free from NaOH, urea and ZnO, which was evidenced from EDX measurements (see ESI, Fig. S3†). Further, washed cellulose beads were subjected to supercritical CO2 drying via ethanol exchange. The diameters of the dried cellulose beads were in the range of 2–2.5 mm. The ZnO concentration was varied as 0, 0.1, 0.2, 0.3, 0.4, 0.5, 1.0, 1.5 & 2.0 wt% during the experiments. The schematic representation of the formation of cellulose beads is depicted in Fig. 1.
 |
| | Fig. 1 Schematic representation of cellulose beads formation. | |
2.3. Characterization of cellulose aerogel microbeads
The supercritical CO2 drying of the wet beads was performed using an autoclave (Eurotechnica, Germany) at 55 °C and 110 bars for 5 days. The size of the cellulose beads were measured using an optical microscope (Zeiss Microscopy, Germany). The spherical beads were randomly selected and placed over a microscopic slide using carbon double tape and the size was calculated by averaging several beads. The skeletal density (ρs) of the beads was measured using AccuPyc II 1340 Gas (Helium) Pycnometer (Micromeritics, Germany). The bulk density (ρb) of the dried beads was calculated by measuring the weight to volume ratio in a 10 mL standard measuring flask. The % porosity (P) of the dried beads were calculated using the following formula, P = {1 − (ρb/ρs)} × 100. Fourier transform infrared spectra of the cellulose bead samples were analyzed using FT-IR, Bruker-Tensor 27 instrument with a resolution of 4 cm−1 and 24 scans over the range of 4000–400 cm−1. For this analysis, the beads were encapsulated in a disc of potassium bromide. XRD measurements were done using D8-Advance X-ray diffractometer from Bruker. The conditions for the measurements are: (i) on the primary side: copper tube with 1600 W (40 mA/40 kV), Göbel mirror (to avoid the effect of peak shift due to geometry of the sample), primary Soller-slit: 2.5° and, (ii) on the secondary side: secondary Soller-slit: 0.2° (nearly parallel beam at the detector). The detector is a Bruker Lynxe-EyeXE (1D-Detector) with a detector opening of 3°. The scanning range from 5 to 85° with a step size of 0.1°. Thermogravimetric analyses of the beads samples were done using Netzsch, TG 209 F1 Iris® instrument with a heating rate of 10 K min−1 under argon atmosphere with a flow of 20 mL min−1 from 25 °C to 800 °C. The surface area, pore volume and pore size distribution of the cellulose beads were measured using N2 adsorption/desorption isotherm analysis using a Micromeritics TriStar II 3020 Gas Sorption Analyzer at 77.3 K temperature with the relative pressure (P/P0) in the range of 0.01–0.1. Before starting analysis, all the samples were outgassed at 130 °C for overnight. The surface area was calculated using the standard Brunauer–Emmett–Teller (BET) method in the relative pressure range 0.01 to 0.1. The pore size distribution plot was obtained from desorption branches using Barrett–Joyner–Halenda (BJH) analysis. The outer and internal structure of cellulose beads were viewed using Merlin type Carl Zeiss scanning electron microscope (SEM) with a low operating voltage of 1–2 kV. All the samples were sputtered with a thin layer of gold. To reveal the internal structure of the beads, they were cut using a sharp blade. The particle size of ZnO was calculated using SEM image by the standard intercept technique.30 The EDX measurements of the dried beads were carried out in LEO 1530 VP with an Oxford Inca EDX-Analyzer (these samples were not gold sputtered).
3. Results and discussion
We first present morphological studies of the cellulose beads, to clearly show, that we did not prepare conventional compact beads, but nanostructured open porous beads, which we call cellulose aerogel beads.
3.1. Morphological studies of the aerogel beads
The morphological changes of the cellulose beads prepared with and without ZnO were analyzed using SEM. The SEM images of the surface of different cellulose beads are depicted in Fig. 2. It can be seen from the images that the spherical morphology of beads is maintained in all cases. All the samples exhibit a porous surface structure consisting of nano-sized fibrils. In the case of the control beads prepared without ZnO (Fig. 2A), the fibrils are very short and continued to merge on the surface because of the slightly smaller solubility of cellulose in NaOH/urea mixture. Whereas in the cases of ZnO loaded samples (Fig. 2B–D), the fibrils are thinner and a fine structure can be seen. As already discussed, the addition of ZnO not only improves the solubility of cellulose but also increases the cellulose molecules amount present in each of the beads, which produces a more dense mesh-like structure on the surface. Similar kind of homogeneous mesh structure was observed for cellulose films prepared with NaOH/urea/ZnO aqueous solutions.20 Moreover, the coagulation of cellulose beads mainly depends on the diffusion of the acid into the beads. Cellulose fibril formation occurs after the first contact with acid medium. It then diffuses into the beads center31 inducing there fibril formation and coagulation. The aerogel beads having a higher amount of cellulose show a more dense structure than that of the controlled beads. Fig. 3 represents the SEM images of fracture surfaces through the cellulose beads prepared with different concentrations of ZnO. These images show that all the cellulose aerogel beads have many pores in their interior structure. The close examination on the fracture surface of control bead shows a short fibril structure which is severely coagulated (0 wt% ZnO). In the cases of beads with ZnO, a more uniform network structure is seen. At 0.5 wt% ZnO concentration, dense presence of a fine fibrillar network structure was resulted. The cross-sectional view shows that the structure was almost uniform in higher concentration of ZnO (0.5 & 1.0 wt%). Moreover, the interior structure of the aerogel beads is composed of interconnected fibrils.
 |
| | Fig. 2 Scanning electron microscopy images of surface of cellulose beads prepared with (A) 0% ZnO, (B) 0.3% ZnO, (C) 0.5% ZnO, and (D) 1.5% ZnO. | |
 |
| | Fig. 3 SEM images of interior cross-sections of cellulose beads with different ZnO concentrations. | |
3.2. FT-IR spectra of cellulose beads
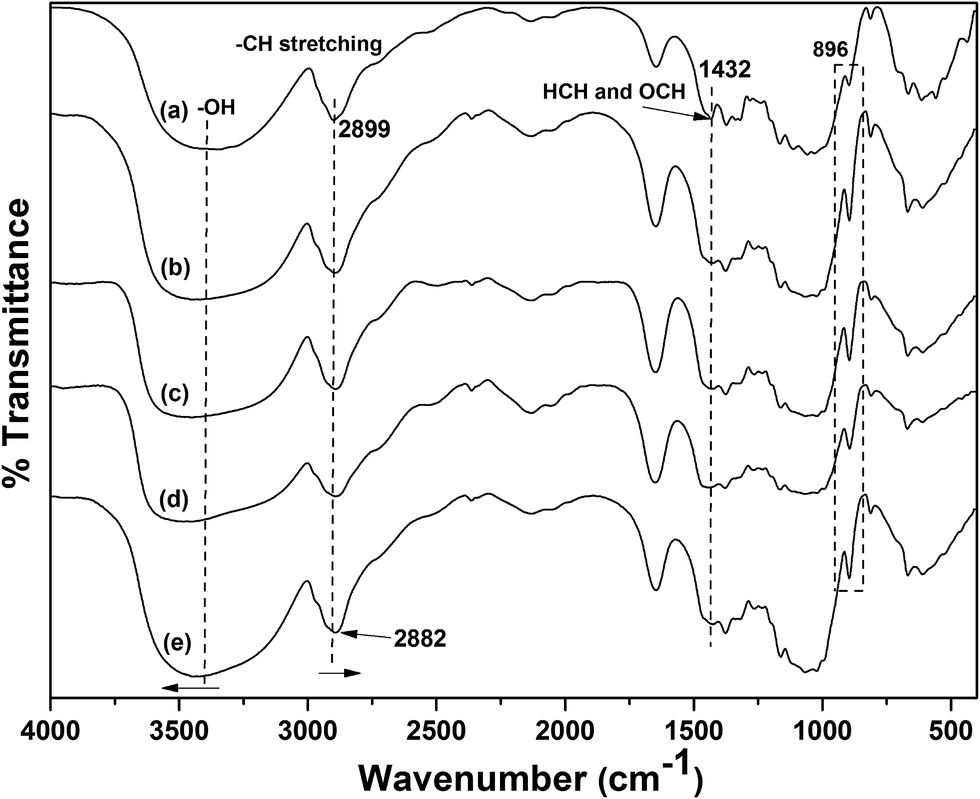
The FT-IR spectra for the commercial cellulose fiber powder and regenerated cellulose beads were given in Fig. 4. The FT-IR spectrum of commercial cellulose fiber powder (Fig. 4a) shows characteristic hydrogen bonded –OH peak in the range of 2990–3700 cm−1. The other characteristic peaks viz., C–H stretching, COO– stretching, HCH and OCH bending vibrations, and C–H deformation appear at 2899, 1645, 1432, and 1374 cm−1, respectively. The peak at 896 cm−1 was assigned to COC, CCO and CCH deformation and stretching vibration of C-5 and C-6 atoms and also C–OH out of plane bending mode observed at 668 cm−1.32 In the cases of cellulose bead samples (i.e., NaOH/urea/ZnO treated samples), there is a slight change in the wavenumber and intensity of certain peaks. The strongly hydrogen bonded –OH peak centered at 3398 cm−1 for original cellulose fiber was shifted to a higher wavenumber of 3443 cm−1 for bead samples as a result of increase in intermolecular hydrogen bonding during regeneration.33 The inter- and intra-molecular bonds present in native cellulose were destructed at the time of dissolution and reorganized during coagulation.33 The C–H stretching peak at 2899 cm−1 was shifted to low wavenumber of 2882 cm−1 (Fig. 4e). This shift may be due to the transformation of cellulose structure during the regeneration process, which affects inter- and intra-molecular hydrogen bonding.34 The peak at 1432 cm−1 from original cellulose mainly attributes to the cellulose I crystal region, which was completely disappeared in the cases of bead samples. This clearly indicates that the dissolution of cellulose in NaOH/urea/ZnO mixture and further regeneration as beads changes the cellulose I structure into cellulose II structure.20 Moreover, the disappearance of 1432 cm−1 peak provides evidence to the destruction of intra molecular hydrogen bonding (formed by O at C6) during regeneration as bead.35 Furthermore, the peak at 896 cm−1 becomes more intense and sharper during regeneration as bead in NaOH/urea/ZnO mixture, which clearly confirms that cellulose I is transformed to cellulose II.32,36
 |
| | Fig. 4 FT-IR spectra of (a) cellulose fiber powder, and cellulose beads with (b) 0% ZnO, (c) 0.1% ZnO, (d) 0.3% ZnO, and (e) 0.5% ZnO. | |
3.3. XRD analyses
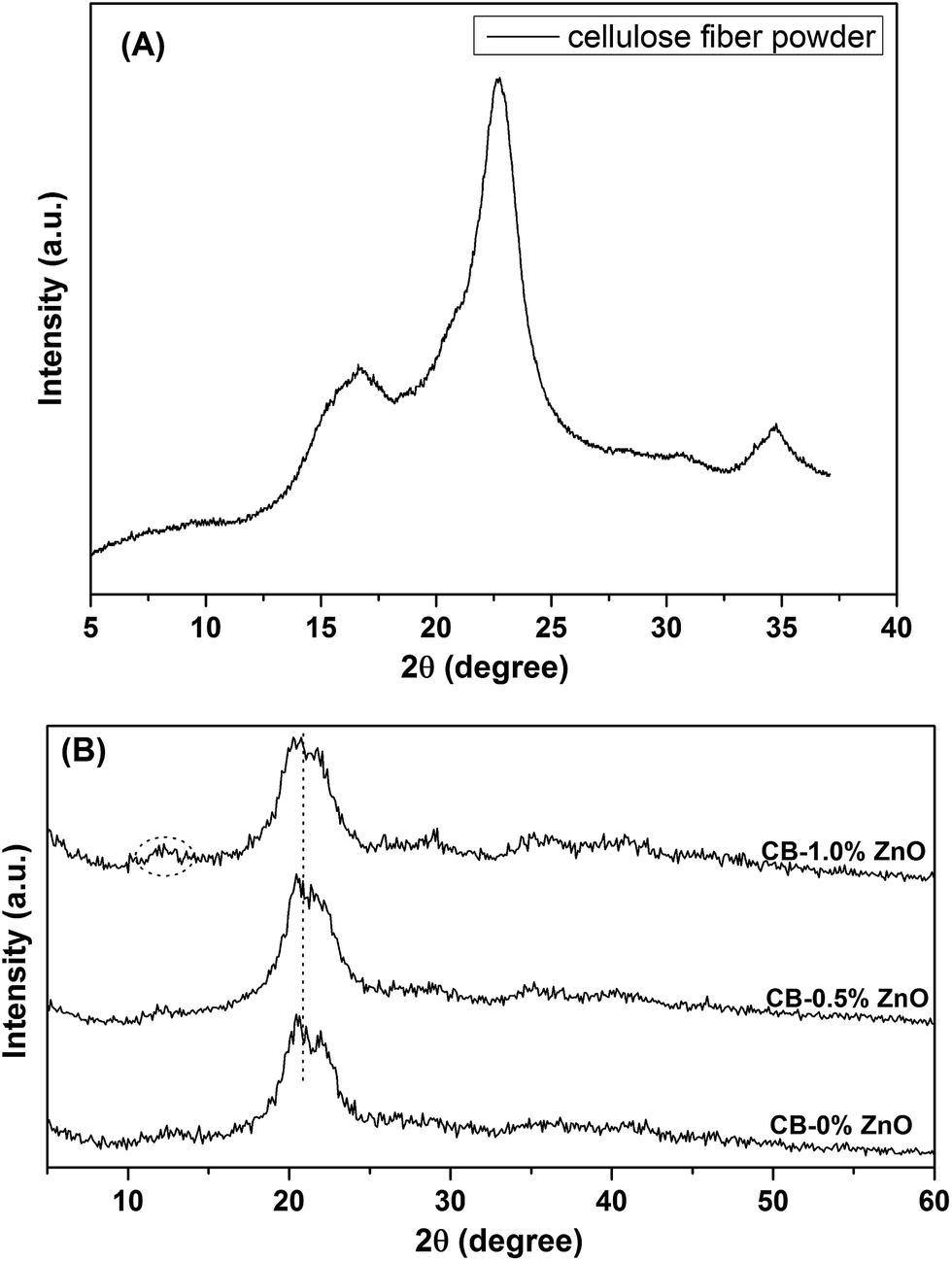
To further prove the conversion of cellulose I to cellulose II form, XRD measurements were done for commercial cellulose fiber powder and regenerated cellulose beads. Their respective XRD patterns are depicted in Fig. 5. XRD pattern of cellulose powder shows three major peaks for the plane at 16.6 (110), 22.6 (200) and 34.8°, which are the characteristics of cellulose I form.37 There is a major change in the XRD pattern of regenerated cellulose beads was observed. The peaks were overlapped and give a broad peak at around 20.9° with peak splitting (two peaks for the planes (110) and (200) centered at 20.2 and 22.1°, respectively). These two peaks are the characteristic of cellulose II form.33,38 There is a low intensity peak at around 12° also characteristic for cellulose II. The peak intensity at 20.9° was increased for the bead samples prepared with ZnO. It can be concluded that the cellulose fiber powder shows the peaks of cellulose I form whereas regenerated beads from NaOH/urea/ZnO mixture shows the characteristic peaks of cellulose II form. This clearly confirms that cellulose structure transformed from cellulose I to cellulose II during dissolution and regeneration in an acid bath.
 |
| | Fig. 5 XRD patterns of (A) cellulose fiber powder, and (B) cellulose beads. | |
3.4. Thermogravimetric analyses of cellulose beads
Thermal stability of the cellulose beads was studied using TGA and their respective TGA and DTG curves were given in Fig. 6 and 7. All aerogel bead samples show similar kind of degradation trend in their respective thermograms. It can be clearly seen from the thermograms that the degradation is governed by three stages. The first stage weight loss is occurred in the range of 25–150 °C, which is mainly due to the removal of moisture as well as adsorbed water present in the cellulose beads system. The percentage weight loss during the first stage was found to be in the range of 6–8% for all the beads samples. In the second stage, the major weight loss occurred between 150 and 500 °C. The second stage degradation is mainly due to the oxidative degradation of the cellulose chain, which ruptures the chain fragments and monolayer present in the cellulose molecules.8,39 In all the cases, the percentage weight loss was estimated at around 74%. This implies that the major part of degradation occurred during the heating of samples between 150 and 500 °C. The thermal degradation above 500 °C is attributed to the elimination of residual carbon. Overall, cellulose beads prepared with ZnO show slightly improved thermal property compared to the beads prepared without ZnO. This can be seen from the residual weight percentage values present at the end of the degradation. The addition of ZnO to the NaOH/urea system provides better dissolution and miscibility of cellulose, which increases the cellulose molecules amount in each of the beads, thereby increasing the residual weight percent at the end.
 |
| | Fig. 6 TGA curves for the prepared cellulose beads. | |
 |
| | Fig. 7 DTG curves for the prepared cellulose beads. | |
3.5. Size and shape analysis of the beads
Overall, the prepared cellulose aerogel beads are clear white and spherical in nature. The images of wet cellulose beads are given in Fig. 8. Their size is in the range of 2.3 to 2.6 mm in diameter. However, after super-critical drying the size of the beads changed. The optical microscopy images of the super critically dried cellulose beads are given in Fig. 9. It can be seen from the microscopic images that their size slightly decreased for the beads prepared with lower concentration of ZnO (0.1 & 0.3 wt%). The reduced size is in the range of 2 to 2.2 mm. However, there is not much reduction in the size of the cellulose beads prepared with higher concentration of ZnO (≥0.5 wt%). The size for the higher concentration of ZnO loaded cellulose beads is in the range of 2.3 to 2.5 mm. Thus the shrinkage occurred during the drying process is controlled by the addition of ZnO. As already mentioned above, the addition of ZnO increases the cellulose molecules amount present in each of the beads, thereby reduces the associated shrinkage during drying process.
 |
| | Fig. 8 Images of the wet cellulose beads prepared without ZnO. | |
 |
| | Fig. 9 Optical microscopy images of the super critically dried cellulose beads. | |
3.6. Density measurements for the beads
Table 1 presents the bulk and skeletal density and the porosity values for the cellulose aerogel beads prepared. All the cellulose bead samples irrespective of ZnO concentration show a skeletal density value of around 1.5 g cm−3. The density value is quite well agreement with the density of bulk cellulose.40 However, there is a different trend observed for the bulk density values. The cellulose aerogel beads prepared with lower concentration of ZnO (0.1–0.4 wt%) exhibit a higher bulk density. This may be due to the shrinkage observed during the drying process for lower ZnO concentration, which occupies more number of beads in the same amount of measuring volume (10 mL measuring flask) compared to the beads with higher ZnO concentration. For higher additions of ZnO, the bulk density values are almost similar with the control cellulose aerogel beads. The porosity of control cellulose aerogel beads was found to be 94%. There is a slight decrease in the porosity values for the beads prepared with lower ZnO concentration (0.1–0.4 wt%). The higher concentrations of ZnO (0.5–2.0 wt%) do not affect the overall porosity of the beads.
Table 1 Density and porosity values for the prepared cellulose beads
| Sample codes |
Skeletal density (g cm−3) |
Bulk density (g mL−1) |
Porosity (%) |
| CB-0% ZnO |
1.51 |
0.082 |
94 |
| CB-0.1% ZnO |
1.49 |
0.181 |
88 |
| CB-0.2% ZnO |
1.49 |
0.182 |
88 |
| CB-0.3% ZnO |
1.48 |
0.226 |
85 |
| CB-0.4% ZnO |
1.48 |
0.250 |
83 |
| CB-0.5% ZnO |
1.48 |
0.089 |
94 |
| CB-1.0% ZnO |
1.47 |
0.083 |
94 |
| CB-1.5% ZnO |
1.47 |
0.095 |
94 |
| CB-2.0% ZnO |
1.51 |
0.088 |
94 |
3.7. BET results of the beads
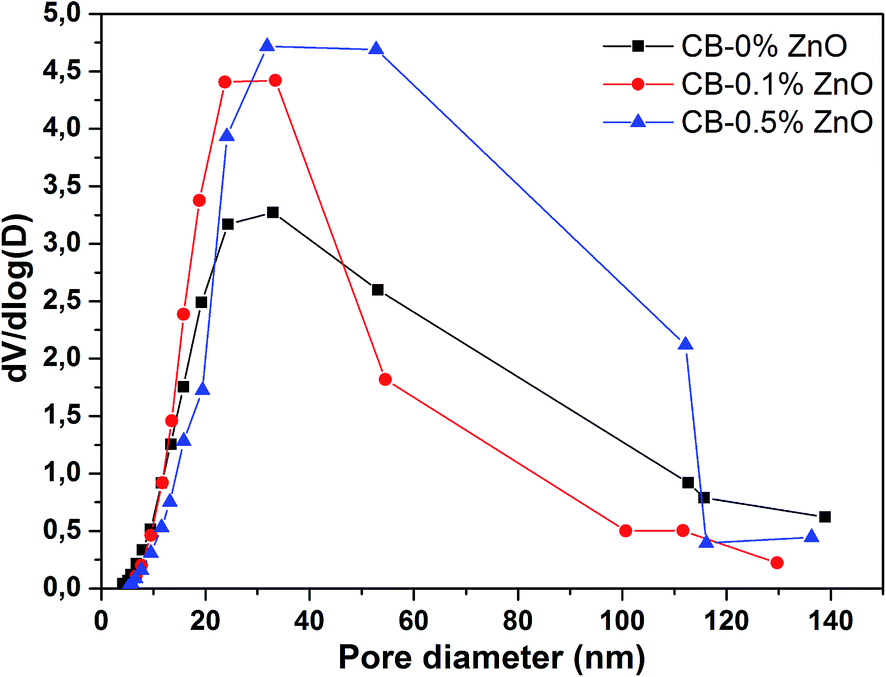
The N2 adsorption/desorption isotherm plots for the dried cellulose aerogel beads are provided in Fig. 10. It can be seen from the isotherms that all cellulose aerogel bead samples exhibit a typical type IV isotherm, which is the characteristic feature of multilayer adsorption on mesoporous solids having strong adsorbent–adsorbate interactions. The BET results for the prepared beads are summarized in Table 2. The surface area (SBET) values for CB-0% ZnO and CB-0.5% ZnO were 341 and 407 m2 g−1, respectively. The surface area of cellulose aerogel bead is greatly improved by a value of 66 m2 g−1 with the addition of 0.5 wt% ZnO to the NaOH/urea mixture. The enhancement of surface area is attributed to the addition of ZnO, since the amphoteric substance forms a stronger hydrogen bonding with the cellulose molecules than in hydrated NaOH. This leads to two effects: first it improves the dissolution of cellulose20 and secondly it keeps the cellulose chains apart from each other due to its large steric hindrance (ZnO exists as Zn(OH)42− in strong alkali).19 During the regeneration process, cellulose molecules reassemble by forming hydrogen bonding together along with the liquid phase. This permits cellulose molecules to occupy more space together with the liquid phase during regeneration, which increases the surface area of cellulose.41 In the cases of lower concentration of ZnO (0–0.3 wt%), the increment in surface area is not quite high. Whereas, there is slightly decreasing trend observed for higher concentration of ZnO (1.0–2.0 wt%). Even though, the average increment of surface area (>40 m2 g−1) in comparison with control cellulose aerogel beads is reasonably good for higher ZnO (0.5–2.0 wt%) loaded bead samples. This may be due to the fact that the overload of ZnO (>0.5 wt%) concentration leads to the decrease of NaOH contribution in cellulose dissolution. Yang et al.19 reported that maximum solubility of cellulose can be achieved by adding a maximum of 0.5 wt% ZnO to the NaOH/urea system. Furthermore, the average pore volume (Vp,avg) of CB-0% ZnO is calculated as 1.48 cm3 g−1. Interestingly, the average pore volume (Vp,avg) values for the cellulose aerogel beads prepared with ZnO show increasing trend irrespective of the concentration of ZnO. The pore size distribution plots for the super-critically dried cellulose beads are given in Fig. 11. The average pore diameter (Davg) for the prepared cellulose beads is about 15–20 nm. Moreover, cellulose beads with 0.5 wt% ZnO show relatively low average pore diameter (15.37 nm) with high surface area of 407 m2 g−1 and an increased pore volume of 1.56 cm3 g−1 compared to a value of 1.48 cm3 g−1 for CB-0% ZnO. It has been reported that pores having smaller size contribute to a higher pore volume, which results in a higher surface area.42 Trygg et al.31 also showed that cellulose beads having higher amounts of smaller sized pores could result in higher specific surface area.
 |
| | Fig. 10 N2 adsorption/desorption isotherm plots for the dried cellulose beads. | |
Table 2 BET results for the prepared cellulose beadsa
| Sample codes |
SBET (m2 g−1) |
Vp,avg (cm3 g−1) |
Vp,ads (cm3 g−1) |
Vp,des (cm3 g−1) |
Davg (nm) |
Dads (nm) |
Ddes (nm) |
| Where, SBET is the surface area obtained from BET, Vp,avg is the average pore volume, Vp,ads is the pore volume from adsorption, Vp,des is the pore volume from desorption, Davg is the average pore diameter, Dads is the pore diameter from adsorption, and Ddes is the pore diameter from desorption. |
| CB-0% ZnO |
341 |
1.48 |
2.34 |
2.39 |
17.39 |
28.83 |
23.61 |
| CB-0.1% ZnO |
344 |
1.72 |
2.47 |
2.57 |
20.09 |
29.25 |
22.65 |
| CB-0.2% ZnO |
352 |
1.87 |
2.69 |
2.76 |
21.25 |
30.76 |
24.70 |
| CB-0.3% ZnO |
346 |
1.76 |
2.31 |
2.39 |
20.40 |
26.51 |
19.45 |
| CB-0.4% ZnO |
323 |
1.61 |
2.05 |
2.15 |
20.04 |
25.51 |
18.12 |
| CB-0.5% ZnO |
407 |
1.56 |
3.42 |
3.50 |
15.37 |
37.89 |
30.59 |
| CB-1.0% ZnO |
399 |
1.61 |
3.41 |
3.49 |
16.12 |
38.14 |
29.84 |
| CB-1.5% ZnO |
387 |
1.67 |
3.28 |
3.36 |
17.28 |
37.08 |
30.06 |
| CB-2.0% ZnO |
377 |
1.83 |
3.28 |
3.38 |
19.45 |
36.63 |
28.82 |
 |
| | Fig. 11 Pore size distribution plots for the super-critically dried cellulose beads. | |
3.8. Speculation on the effect of ZnO
The results suggest to us another understanding of the action of ZnO on the dissolution, aggregation and gelation behavior of cellulose in NaOH/urea mixtures. These ideas are somehow speculative and shall be taken as a first hypothesis. In contrast to the literatures cited above, we think that several processes occur in NaOH/urea mixtures at a pH of around 14. First of all ZnO is dissolved in sodium hydroxide solution by forming Zn(OH)42− according to Degen and Kosec.43 The solubility of ZnO in NaOH mixtures is, however, limited and thus after the solubility limit is reached, which is around 0.5 wt% ZnO, precipitates stay in the solution. If the average particle size of the ZnO particles was initially before adding them to the solution R0, then the dissolution process leads to a reduction in average particle size (the smaller ones are completely dissolved and the bigger ones in the initial size distribution become smaller) to a value R≪R0. These left-over ZnO particles are negatively charged at their surface and are covered by a layer of sodium-zincate.43 The amount of free zincate in the solution increases first upon the addition of ZnO-particles but after the maximum solubility is reached, it decreases again.
Since ZnO has an effect on the dissolution and aggregation of cellulose in NaOH/urea mixtures and ZnO is dissolved by forming zincate molecules in solution, the zincate molecules must have an effect on the dissolution itself. We do not think that their main effect is attracting water. One could imagine the following picture: in the amorphous regions of cellulose, both NaOH and zincate diffuse, leading there to a local swelling and stretching thereby intermolecular hydrogen bonds in the adjacent crystalline areas are weakened. The stress exerted on these bonds is proportional to the swelling. The proportionality constant should be the compressibility of the amorphous regions. The extent of swelling depends on the molecule size or volume. NaOH/water would lead then to a smaller swelling compared to the additional action of the much larger zincate molecules. The stretching of the intermolecular hydrogen bonds in the crystalline areas reduces the bond energy and thus eventually breaks these bonds leading to free cellulose molecules, which are complexed at position 6 of the pyranose ring by zincate/urea. If the solubility limit of ZnO in the given solution is reached, the amount of zincate being able to diffusive into amorphous regions decreases and thus the potency of the solution to further dissolve cellulose is reached and decreases in as much as ZnO-precipitates attract sodium zincate at their surface.43
The ability of the complexed cellulose molecules to (re)-aggregate is reduced, since the Brownian mobility depends on the length and the slenderness ratio of a molecule.44 A rough calculation shows that the mobility is reduced like the logarithm of the square root of the ratio of the molar volume of cellulose to complexed cellulose. We calculate a factor of up to 2 for the reduction in mobility. This would mean that the aggregation due to translational or rotational Brownian motion of cellulose molecules with attracted zincate/urea complexes is impeded and that the aggregation time increases. This would, however, essentially affect the formation of nano-fibrils and not the network formation of such fibrils leading finally to gelation. Gelation needs additional considerations especially in NaOH/urea solution, since gelation is induced by neutralization of the NaOH solution. Details of these model calculations are currently worked out and will be published soon.
4. Conclusion
In the present work, we prepared cellulose aerogel microbeads by dissolving cellulose fiber powder in NaOH/urea/ZnO aqueous solution and coagulating in hydrochloric acid medium. The variation of ZnO concentration on the structure and properties of cellulose beads was studied using different analytical techniques like, FT-IR spectroscopy, thermogravimetric analysis, N2 adsorption/desorption isotherm, and scanning electron microscopy. Cellulose aerogel beads prepared with lower concentration of ZnO (0.1–0.4 wt%) show shrinkage after super critical CO2 drying. Beads with higher ZnO content (0.5–2.0 wt%) almost retain their original size even after drying. Cellulose beads prepared with 0.5 wt% ZnO show higher surface area of 407 m2 g−1 with large porosity (94%) and increased pore volume of 1.56 cm3 g−1 compared to control cellulose beads (1.48 cm3 g−1). SEM images show that all aerogel beads exhibit good spherical shape with homogeneous pore structure on the surface. These results indicate that the bulk density, surface area, pore volume and pore size of the cellulose aerogel beads can be tuned by changing the concentration of ZnO.
Acknowledgements
One of the authors (K. Seeni Meera) wishes to thank DLR-DAAD post-doctoral fellowship programme for the financial assistance. We wish to thank Dr Matthias Kolbe, DLR for his assistance with SEM analyses.
References
- M. J. John and S. Thomas, Carbohydr. Polym., 2008, 71, 343–364 CrossRef CAS PubMed.
- C. Salas, T. Nypelo, C. Rodriguez-Abreu, C. Carrillo and O. J. Rojas, Curr. Opin. Colloid Interface Sci., 2014, 19, 383–396 CrossRef CAS PubMed.
- X. Y. Qiu and S. W. Hu, Materials, 2013, 6, 738–781 CrossRef CAS PubMed.
- M. Gericke, J. Trygg and P. Fardim, Chem. Rev., 2013, 113, 4812–4836 CrossRef CAS PubMed.
- R. Sescousse, R. Gavillon and T. Budtova, J. Mater. Sci., 2011, 46, 759–765 CrossRef CAS.
- M. Hirota, N. Tamura, T. Saito and A. Isogai, Cellulose, 2009, 16, 841–851 CrossRef CAS.
- K. Thümmler, S. Fischer, A. Feldner, V. Weber, M. Ettenauer, F. Loth and D. Falkenhagen, Cellulose, 2011, 18, 135–142 CrossRef.
- X. M. Zhang, H. W. Yu, H. J. Yang, Y. C. Wan, H. Hu, Z. Zhai and J. M. Qin, J. Colloid Interface Sci., 2015, 437, 277–282 CrossRef CAS PubMed.
- J. Lindh, D. O. Carlsson, M. Strømme and A. Mihranyan, Biomacromolecules, 2014, 15, 1928–1932 CrossRef CAS PubMed.
- T. Heinze and T. Liebert, Prog. Polym. Sci., 2001, 26, 1689–1762 CrossRef CAS.
- H. Jin, Y. Nishiyama, M. Wada and S. Kuga, Colloids Surf., A, 2004, 240, 63–67 CrossRef CAS PubMed.
- K.-F. Du, M. Yan, Q.-Y. Wang and H. Song, J. Chromatogr. A, 2010, 1217, 1298–1304 CrossRef CAS PubMed.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- S. Hoepfner, L. Ratke and B. Milow, Cellulose, 2008, 15, 121–129 CrossRef CAS.
- L. Ratke, in Aerogels Handbook, ed. M. A. Aegerter, N. Leventis and M. M. Koebel, Springer, New York, 2011, ch. 9, pp. 173–190, DOI:10.1007/978-1-4419-7589-8_9.
- J. Zhou and L. Zhang, Polym. J., 2000, 32, 866–870 CrossRef CAS.
- B. Xiong, P. Zhao, K. Hu, L. Zhang and G. Cheng, Cellulose, 2014, 21, 1183–1192 CrossRef CAS.
- J. Cai, L. N. Zhang, J. P. Zhou, H. S. Qi, H. Chen, T. Kondo, X. M. Chen and B. Chu, Adv. Mater., 2007, 19, 821–825 CrossRef CAS PubMed.
- Q. L. Yang, H. S. Qi, A. Lue, K. Hu, G. Z. Cheng and L. N. Zhang, Carbohydr. Polym., 2011, 83, 1185–1191 CrossRef CAS PubMed.
- Q. L. Yang, X. Z. Qin and L. N. Zhang, Cellulose, 2011, 18, 681–688 CrossRef CAS.
- M. Kihlman, B. F. Medronho, A. L. Romano, U. Germgard and B. Lindman, J. Braz. Chem. Soc., 2013, 24, 295–303 CrossRef CAS.
- W. Q. Liu, T. Budtova and P. Navard, Cellulose, 2011, 18, 911–920 CrossRef CAS.
- P. Navard, F. Wendler, F. Meister, M. Bercea and T. Budtova, in The European Polysaccharide Network of Excellence (EPNOE), ed. P. Navard, Springer, Vienna, 2013, ch. 5, pp. 91–152, DOI:10.1007/978-3-7091-0421-7_5.
- A. Demilecamps, G. Reichenauer, A. Rigacci and T. Budtova, Cellulose, 2014, 21, 2625–2636 CrossRef CAS.
- X. Xiong and J. Duan, in The Role of Green Chemistry in Biomass Processing and Conversion, John Wiley & Sons, Inc., 2012, pp. 205–240, DOI:10.1002/9781118449400.ch6.
- F. Y. Fu, L. Y. Li, L. J. Liu, J. Cai, Y. P. Zhang, J. P. Zhou and L. N. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 2597–2606 CAS.
- Y. Jiang, Y. Song, M. Miao, S. Cao, X. Feng, J. Fang and L. Shi, J. Mater. Chem. C, 2015, 3, 6717–6724 RSC.
- I. Karadagli, B. Schulz, M. Schestakow, B. Milow, T. Gries and L. Ratke, J. Supercrit. Fluids, 2015 DOI:10.1016/j.supflu.2015.06.011.
- L. S. Blachechen, P. Fardim and D. F. S. Petri, Biomacromolecules, 2014, 15, 3440–3448 CrossRef CAS PubMed.
- E. E. Underwood, Quantitative stereology, Addison-Wesley Pub. Co., 1970 Search PubMed.
- J. Trygg, P. Fardim, M. Gericke, E. Makila and J. Salonen, Carbohydr. Polym., 2013, 93, 291–299 CrossRef CAS PubMed.
- S. Y. Oh, D. I. Yoo, Y. Shin and G. Seo, Carbohydr. Res., 2005, 340, 417–428 CrossRef CAS PubMed.
- X. G. Luo and L. N. Zhang, J. Chromatogr. A, 2010, 1217, 5922–5929 CrossRef CAS PubMed.
- S. Y. Oh, D. I. Yoo, Y. Shin, H. C. Kim, H. Y. Kim, Y. S. Chung, W. H. Park and J. H. Youk, Carbohydr. Res., 2005, 340, 2376–2391 CrossRef CAS PubMed.
- L. N. Zhang, D. Ruan and J. P. Zhou, Ind. Eng. Chem. Res., 2001, 40, 5923–5928 CrossRef CAS.
- D. S. Zhao, M. S. Liu, H. W. Ren, H. Li, L. L. Fu and P. B. Ren, Fibers Polym., 2013, 14, 1261–1265 CrossRef CAS.
- N. Sun, R. P. Swatloski, M. L. Maxim, M. Rahman, A. G. Harland, A. Haque, S. K. Spear, D. T. Daly and R. D. Rogers, J. Mater. Chem., 2008, 18, 283–290 RSC.
- A. John, H. U. Ko, D. G. Kim and J. Kim, Cellulose, 2011, 18, 675–680 CrossRef CAS.
- H. L. Cai, S. Sharma, W. Y. Liu, W. Mu, W. Liu, X. D. Zhang and Y. L. Deng, Biomacromolecules, 2014, 15, 2540–2547 CrossRef CAS PubMed.
- M. Ettenauer, F. Loth, K. Thümmler, S. Fischer, V. Weber and D. Falkenhagen, Cellulose, 2011, 18, 1257–1263 CrossRef CAS.
- R. Gavillon and T. Budtova, Biomacromolecules, 2008, 9, 269–277 CrossRef CAS PubMed.
- P. R. Aravind, L. Ratke, M. Kolbe and G. D. Soraru, J. Sol-Gel Sci. Technol., 2013, 67, 592–600 CrossRef CAS.
- A. Degen and M. Kosec, J. Eur. Ceram. Soc., 2000, 20, 667–673 CrossRef CAS.
- J. K. G. Dhont and W. J. Briels, Rod-Like Brownian Particles in Shear Flow, Sections 3.10–3.16, in Soft Matter, Volume 2. Complex Colloidal Suspensions, ed. G. Gompper and M. Schick, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2015 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: EDX spectra of cellulose beads and SEM & PSD of ZnO. See DOI: 10.1039/c5ra17366c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Barbara Milow and
Lorenz Ratke
,
Barbara Milow and
Lorenz Ratke