
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Processing and characterization of large diameter ceramic SiCN monofilaments from commercial oligosilazanes†
O.
Flores
a,
R. K.
Bordia
b,
S.
Bernard
c,
T.
Uhlemann
a,
W.
Krenkel
a and
G.
Motz
*a
aCeramic Materials Engineering, University of Bayreuth, D-95447 Bayreuth, Germany. E-mail: guenter.motz@uni-bayreuth.de
bDepartment of Materials Science and Engineering, Clemson University, SC 29634-0971, Clemson, USA
cIEM (Institut Europeen des Membranes), UMR 5635 (CNRS-ENSCM-UM), Universite Montpellier, Place E. Bataillon, F-34095, Montpellier, France
First published on 9th December 2015
Abstract
This work reports the processing of large diameter ceramic SiCN monofilaments via the precursor route using two chemically different polysilazanes ML33S and HTTS self-synthesized from respective commercially available oligosilazanes. The melt-spinning of continuous polymer fibers with controllable diameters from 35 to 150 μm and their pyrolysis to ceramic SiCN fibers is not influenced by differences in the chemical structure of the polysilazanes. In contrast, the necessary e-beam curing dose is reduced by Si-vinyl groups from 600 kGy for ML33S (vinyl free) to 200 kGy for HTTS derived polymer fibers. The curing step leads to an enhanced handleability important for further pyrolysis at 1100 °C in nitrogen and to an increase in ceramic yield. The resulting ceramic SiCN fibers from both systems have similar mechanical and thermal behavior, indicating quite a low influence of the polysilazane type on these properties. For the first time a comprehensive investigation of the effect of fiber diameter on the tensile strength is reported for SiCN fibers. The average strength increases from ∼800 MPa for 90 μm diameter fibers to ∼1600 MPa for the 30 μm diameter fibers. Bend Stress Relaxation (BSR) tests demonstrated that no stress relaxation occurs up to 1000 °C for SiCN monofilaments and the creep resistance is equal to or better than commercially available SiC monofilaments produced by chemical vapour deposition (CVD). The oxidation resistance is also comparable to commercially available oxygen free CVD SiC fibers (SCS-6). The ceramic fibers in this study were pyrolyzed at low temperature (1100 °C) and have high oxygen content (13 to 29 wt%). The high temperature creep resistance and oxidation resistance is expected to improve if the oxygen content is reduced and the pyrolysis temperature increased.
1. Introduction
Lightweight thermostructural materials are needed in next generation aero turbine engine components to improve the energy efficiency due to weight reduction and increase of the operating temperature. They are also needed for other advanced applications including heat exchangers, rocket propulsion, fusion reactors and UHTC parts for supersonic and hypersonic vehicles. In these applications, ceramic fibers play an important role as reinforcements of metallic and ceramic matrices to realize the manufacturing of composite materials with low density and for high temperature applications.Research activities in the field of high performance non-oxide ceramic fibers led, over the last 50 years, to the development of commercially available chemical vapour deposited (CVD) SiC monofilaments1–3 and three generations of SiC fibers based on polymer derived ceramics (PDCs).2,4–6 These high performance fibers comprise not only superior mechanical properties, such as tensile strength > 2 GPa and stiffness, but also thermal stability at temperatures higher than 800 °C, which are essential for the reinforcement of materials based on metallic and ceramic matrix composites (MMCs and CMCs respectively).
For MMCs applications, such as combustion engines and high efficiency turbines, titanium matrix composites (TMCs) reinforced with CVD SiC monofilaments are promising candidates, as they combine the high strength, stiffness and creep resistance of SiC monofilaments with the high ductility, toughness and corrosion resistance of titanium alloys.7,8 Because SiC fibers are generally reactive with titanium alloys at high temperatures, fabrication processes that allow a controlled chemical reaction with the metal matrix, mainly dominated by diffusion, enables the formation of a strong fiber–matrix interface and good adhesion. This good adhesion is needed for the efficient load transfer from the metal matrix to the SiC fibers. The major obstacle for the application of TMCs in the industry is the high material cost of monofilament SiC fibers. They are among the most expensive ceramic fibers with prices up to ∼8000 € per kg (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U$ per kg).2,8,9
000 U$ per kg).2,8,9
In contrast, the continuous processing of fine diameter fibers from inorganic polymers is still the most attractive method for the manufacturing of cost-effective non-oxide ceramic fibers. Ceramic fibers from SiCN systems, such as polysilazanes, comprise good oxidation stability up to ∼1500 °C (due to the nitrogen content) and corrosion resistance in acids and bases. Beside this they offer a remarkable cost reduction, due to relatively inexpensive precursors, use of low e-beam doses for their curing and relatively simple processing.2,10,11
In this paper we report on the processing and properties of large diameter SiCN monofilaments made from preceramic polymers as an alternative to CVD SiC monofilaments. Two different types of meltable polysilazanes with tailored properties were recently developed in our chair.12,13 These polymers open new opportunities for cost effective processing of ceramic SiCN fibers with larger diameters as demonstrated in this investigation.
Therefore, the aim of this work was to melt spin tailored polysilazanes fibers of large diameter, to investigate the influence of the electron beam irradiation dose on the curing of melt spun green fibers and to study the pyrolysis of these green polymeric fibers to ceramic fibers. Finally, the mechanical and thermal properties, including strength, strength distribution, creep resistance and oxidation behavior, of these fibers have been investigated and compared to commercially available SiC fibers.
2. Experimental procedure
Both polymers, ML33S and HTTS, used for the processing of ceramic SiCN fibers are produced by a selective chemical cross-linking of two commercially available liquid oligosilazanes ML33 and HTT1800, respectively, which were purchased from Clariant Advanced Materials GmbH (Sulzbach, Germany) (Fig. 1). | ||
| Fig. 1 Simplified chemical structures of the liquid ML33 and HTT1800 oligosilazanes. | ||
Both tailored polymers are meltable solids, having suitable viscoelasticity and thermal stability up to 170 °C for the melt-spinning of fibers. More details about the synthesis and characterization of ML33S and HTTS, including their rheology and thermal stability, have been reported elsewhere.12–14
2.1. Processing of ceramic SiCN monofilaments
Monofilaments from ML33S or HTTS polysilazanes were melt-spun in a lab-scale gas pressure melt spinning equipment. About 8 g of the polymer were filled in the vessel of the spin equipment, melted at temperatures between 110 and 130 °C, evacuated to remove gas bubbles and left at the desired temperature for 15 minutes to ensure a constant and uniform temperature. Afterwards, nitrogen over pressure was applied to allow the melt-spinning of filaments through a spinneret followed by winding of the fibers. Monofilaments with diameter > 70 μm were melt spun through a spinneret with a single capillary having a diameter of 800 μm. To investigate the dependence of the strength of the ceramic fibers on the diameter, filaments with diameter < 70 μm were also melt spun by using a spinneret with 7 holes, each one with a diameter of 400 μm.For the investigation of the electron beam irradiation dose needed to render the precursor unmeltable, cylindrical samples having diameter of 20 mm and thickness of 10 mm were prepared from ML33S or HTTS polysilazanes by melting the polymer in nitrogen atmosphere in a silicone mold. The samples were packed under nitrogen atmosphere in a polyethylene (PE) plastic sheet and sent to the company Beta-Gamma-Service GmbH, Saal, Germany. An acceleration voltage of 10 MeV and doses up to 1000 kGy were applied for the electron beam curing experiments of the samples.
The gel fraction of the precursors after irradiation was determined based on the German standard DIN 16892.15 About 0.25 g of the cured sample powder was solved in toluene and refluxed for 4 hours at 80 °C. After filtration through a regenerated cellulose filter with a pore size of 0.2 μm, the undissolved fraction remaining on the filter was dried and weighed for the calculation of the gel fraction according to the following equation:
 | (01) |
For the investigation of the transition from the polymer to amorphous ceramic SiCN material thermal gravimetric analysis (TGA) was performed, using a Linseis L81 A1550 unit (Linseis, Germany). Approximately 10 mg of the sample was heated from 25 to 1300 °C with a heating rate of 5 K min−1 in nitrogen atmosphere. The ceramic yield was calculated from the mass change of the material after the thermal treatment.
Based on the curing results obtained from the cylindrical samples, the collected monofilaments were cured at BGS GmbH, Saal, with an acceleration voltage of 10 MeV and doses up to 600 kGy. After curing, the fibers were converted into amorphous ceramic SiCN fibers by continuous pyrolysis in a tubular furnace (model GERO Gero FA100-500/13) equipped with a bobin at each end of the furnace tube. The pyrolysis was conducted at a maximum temperature of 1100 °C in nitrogen atmosphere. Cured green fibers were continuously pulled through the furnace using a fiber winding unit with a velocity of 0.3 cm min−1. The elemental composition of the uncured, cured and of the ceramic SiCN fibers was analysed at Pascher Microanalytical Laboratory (Remagen, Germany).
2.2. Properties of ceramic SiCN monofilaments
Tensile tests were carried out for both the cured green and ceramic SiCN monofilaments at ambient conditions using a universal mechanical testing machine (Inspekt Mini, Hegewald & Peschke GmbH, Germany). From each batch 40 samples were selected and their tensile strength was determined. According to DIN-ENV 1007-4 individual filaments were glued onto a split cardboard frame with a gauge length of 25 mm.16 A 20 N load cell was used with a cross-head speed of 4 mm min−1. Fiber diameters were determined prior to the mechanical test with an optical microscope (Zeiss, Axiotech 100) equipped with a camera for digital analysis of the picture. After the tensile test, filaments that broke in the extremities of the split cardboard frame were excluded from the analysis.In order to measure the creep resistance of the amorphous ceramic SiCN fibers, the Bend Stress Relaxation (BSR) method developed by Morscher et al.17 was used for single monofilaments with diameter > 70 μm. Fibers were tied into a loop with a radius R0 of 10 mm at room temperature and heated up in air or nitrogen atmosphere for 1 hour to temperatures ranging from 900 to 1200 °C. After the thermal treatment of the sample, the fiber loop was cut at room temperature and the resulting radius of curvature Ra measured. To have a quantitative measurement of the creep resistance of the fibers, the BSR ratio (mBSR(T, t) = 1 − R0/Ra) was calculated. For the evaluation of mBSR, 4 samples of each ceramic SiCN fiber charge were used for each test condition. The mean value of the mBSR of these samples was used for the discussion.
The oxidation resistance of the ceramic SiCN fibers was investigated using the TGA equipment (Linseis, L81/1550), and compared with commercial ceramic SiC fibers, type SCS 6. Monofilaments were cut in samples of 2 cm in length and placed in an alumina crucible. The samples were heated up to a maximum temperature ranging from 900 to 1300 °C, heating rate of 10 K min−1 and a dwell time up to 12 hours under flowing oxygen (5 L min−1). After oxidation, the morphology and thickness of the oxide layer on the fiber was analysed by scanning electron microscopy (SEM, Zeiss, 1540EsB) coupled with an Energy Dispersive X-ray Spectrometer (EDS, Thermo Scientific, NORAN System 6).
3. Results and discussion
3.1. Processing of polymer derived ceramic SiCN fibers
Based on the molecular, thermal and rheological analysis of the polysilazanes manufactured by controlled cross-linking,12,13 continuous green fibers were processed in air, using the lab-scale melt spinning equipment. After the green fibers were spun in air and left for 3 hours on the spool in contact with air, the oxygen content of the fibers remains <0.5 wt%. The oxygen incorporation is due to the reaction between the surface of the fiber with the moisture in the environment18 as described by Hacker,11 who used the self-synthesized ABSE polysilazane for processing of ceramic SiCN fibers. Fig. 2 shows a large diameter melt spun monofilament wound on a spool. | ||
| Fig. 2 (a) Melt spinning of polymer green monofilament and a spool with wound monofilaments. SEM micrographs of (b) the cross-section and (c) the surface of ML33S derived green monofilaments. | ||
For the processing of green fibers, based on prior knowledge in our laboratory, a viscosity of 5 × 103 Pa s was chosen to ensure a continuous viscoelastic flowability of the polymer. Based on rheological measurements, this required a processing temperature of about 50 K higher than the glass temperatures of the polymers.12,13 In addition to the viscosity and the spinneret hole diameter, the final diameter of the green fibers is controlled by the pull-off speed. Polymer monofilaments having diameters between ∼35 and 150 μm were successfully processed by adjusting the temperature (and hence the viscosity of the melt) between 110 and 120 °C and by varying the pull-off speed between 10 and 30 m min−1.
The e-beam irradiation is not only crucial to turn the green fibers into infusible solids, but also improves their flexibility and handleability for further pyrolysis into ceramic fibers.
Commercially available SiC fibers require high electron beam doses, between 7500 and 20000 kGy.19 The irradiation dose to cross-link Hi-Nicalon fibers from polycarbosilane (PCS) can be reduced by the addition of vinyl sources, such as polyvinylsilane.20,21 But the work about the influence of the irradiation dose on the cross-linking behavior of polyorganosilazanes is rather limited.22,23
Therefore the present investigations to determine the minimum e-beam dose are based on scientific works regarding the development of ceramic SiCN fibers from ABSE polysilazane.22 In order to quantify the curing results, the gel content of the polymer ML33S and HTTS fibers after irradiation was measured (using eqn (01)).
Fig. 3 demonstrates that the curing behavior of the polysilazanes strongly depends on their functional groups. Due to the absence of Si-vinyl groups in ML33S, the required dose for its curing is higher than that for HTTS. Hence, an e-beam dose ≥600 kGy is required to reach a gel content of about 60% and to render the ML33S green fibers infusible. Melting tests performed with the polymers after curing confirm that ML33S samples treated with e-beam doses greater than 600 kGy maintain their shape (remain infusible).
 | ||
| Fig. 3 Gel content of ML33S and HTTS polysilazanes after curing with electron beam doses up to 1000 kGy and comparison with the melting test of the respective cured samples. | ||
HTTS already reaches a gel content of nearly 100% after treatment with a low dose of only 200 kGy due to the highly reactive Si-vinyl groups. In contrast to the curing of ML33S, all HTTS samples showed a yellowish appearance, which is probably a sign of the formation of reactive free radicals or conjugated bondings. The melting test confirmed that all cured HTTS samples were already infusible even after treating with a very low dose of 200 kGy, which is enough to obtain infusible HTTS green fibers.
With the additional cross-linking of the precursors by using e-beam irradiation, their ceramic yield slightly increases due to the further reduction of volatile oligomers. In Fig. 4a the TGA curve indicates that a dose of only 200 kGy to cure ML33S has small influence on the reduction of released volatile oligomers, occurring between 200 and 400 °C. However with doses ≥400 kGy this reduction is more pronounced and the ceramic yield for cured ML33S materials increases to up to ∼78 wt% in comparison to a ceramic yield of 70 wt% for the uncured ML33S. However the increase in the ceramic yield for the uncured (78 wt%) to the e-beam cured HTTS samples (∼82 wt%) is not significant (Fig. 4b).
 | ||
| Fig. 4 The dependence of mass change during pyrolysis on the e-beam dose of (a) ML33S and (b) HTTS polysilazanes. | ||
These results clearly illustrate that the Si-vinyl groups reduce the necessary electron beam dose for the curing of HTTS derived green fibers significantly in comparison to the less reactive ML33S derived green fibers. Furthermore, while both types of melt spun uncured green fibers are brittle, especially the cured green fibers from HTTS appear more flexible and stronger, which is important for the subsequent pyrolysis process. After exposure to an e-beam dose of 600 kGy, cured green fibers from HTTS have a mean tensile strength of 65 MPa, while cured ML33S derived green fibers reached a mean strength of only 43 MPa.
Concerning the continuous pyrolysis of the cured fibers into ceramic fibers, a minimum diameter for the take-up spool used to pull the ceramic fibers leaving the furnace was chosen to avoid that the fibers with d > 70 μm break on the spool. For this purpose, prior to the continuous pyrolysis, about 1 meter of the cured monofilaments with diameter ∼ 100 μm were cut in samples with 20 cm length, deposited on a graphite boat and pyrolysed in a furnace at 1000 °C in nitrogen atmosphere for 1 hour. The pyrolysed monofilaments were bent until they broke and the mean value of the bending diameter prior to the breaking was taken as the minimum spool diameter used to pull the SiCN fibers.
After the continuous pyrolysis in a horizontal tubular furnace in nitrogen atmosphere the resulting ceramic SiCN fibers leave the furnace passing through the guide pulley before they are finally pulled on a spool with a diameter of 160 mm by a motor operated coiling unit (Fig. 5a and b). Fig. 5b shows a bundle of 3 pyrolysed ceramic monofilaments with a length of more than 10 meters on the spool.
 | ||
| Fig. 5 (a) Ceramic SiCN fibers from HTTS with diameter of ∼100 μm leaving the tubular furnace and (b) continuously pulled on the spool. | ||
For all investigated fiber batches continuous pyrolysis lead to amorphous ceramic SiCN fibers with regular and smooth surface. SEM micrograph of HTTS derived ceramic fibers confirms diameters up to 100 μm, with a dense core and without noticeable defects or pores (Fig. 6). Due to the similarity, micrographs of ML33S derived ceramic fibers are not shown.
 | ||
| Fig. 6 SEM micrographs of (a) the cross section and (b) surface of amorphous ceramic SiCN fibers from HTTS. | ||
The oxygen content of the processed amorphous ceramic SiCN fibers is similar or higher than for Nicalon fibers (Table 1). Although the oxygen content of the uncured polymers is only <0.5 wt%, after their melt-spinning in air and curing with electron beam irradiation in inert atmosphere it increases to 3.5 and 4.7 wt% for ML33S and HTTS derived green fibers, respectively. After pyrolysis in nitrogen atmosphere the oxygen content of ML33S derived ceramic fibers reaches ∼13 wt% while HTTS derived ceramic fibers contain ∼29 wt% oxygen. This high increase in oxygen content is due to the handling and long exposure of the cured fibers in air before they are pulled through the furnace at a very low velocity of 0.3 cm min−1, as described in Chapter 2.1.
| Structure | Si (wt%) | C (wt%) | N (wt%) | O (wt%) | H (wt%) | Empirical formula | |
|---|---|---|---|---|---|---|---|
| ML33S | Uncured polymer | 43.7 | 24.0 | 23.8 | 0.4 | 8.1 | SiC1.28N1.09H5.15 |
| Cured polymer | 44.0 | 23.6 | 21.7 | 3.5 | 7.2 | SiC1.25N0.99O0.14H4.55 | |
| Ceramic | 53.0 | 15.7 | 18.4 | 12.9 | <0.05 | SiC0.69N0.70O0.43 | |
| HTTS | Uncured polymer | 42.4 | 26.2 | 23.3 | 0.4 | 7.6 | SiC1.44N1.1H5.02 |
| Cured polymer | 42.1 | 25.8 | 20.5 | 4.7 | 6.9 | SiC1.43N0.98O0.20H4.56 | |
| Ceramic | 46.7 | 19.2 | 5.5 | 28.6 | <0.03 | SiC0.96N0.23O1.07 | |
| Nicalon2 | Ceramic | 57 | 32 | — | 12 | — | — |
During exposure in air, the nitrogen of the silazanes is replaced by oxygen.18 This fact becomes evident especially for HTTS derived fibers as a consequence of the higher content of moisture sensitive Si–H groups. Beside this, vinyl groups are present in this precursor, which are activated during e-beam curing, distinguishable on the change in color from colorless to yellowish due to the formation of reactive radicals. Clearly, this enhanced reactivity also leads to an additional formation of Si–O bondings.21,22 More details about the influence of oxygen content in the creep and oxidation stability of the processed SiCN fibers will be later on discussed in Chapters 3.2.2 and 3.2.3.
3.2. Mechanical and thermal properties of ceramic SiCN fibers
Fig. 7 shows the tensile strength of ML33S and HTTS derived ceramic SiCN fibers as a function of the fiber diameter. In the case of ML33S derived ceramic fibers, there is significant scatter in strength for fibers with a diameter between 40 and 50 μm, but the tendency to higher strength values with the reduction of the diameter is clearly illustrated. Such scatter results from the not optimized melt-spinning process. With this simple lab-scale gas pressure melt spinning equipment it is impossible to remove all gas bubbles from the polymer melt, which lead to pores within the resulting fibers. However, especially for HTTS derived ceramic fibers the effect of fiber diameter on tensile strength is much clear due to lower scatter. For both ceramic fiber types the tensile strength values are similar for the same diameter. While fibers with about 100 μm have a tensile strength of ∼0.8 GPa, thinner fibers with a diameter of 30 μm have a mean tensile strength as high as ∼1.5 GPa. A further reduction of the fiber diameter should lead to tensile strength values higher than 2 GPa. For comparison, the tensile strength of commercially available SCS-6 fibers is about 3.9 GPa.2 This very high value is a result of the CVD process, which leads to nearly defect free SiC fibers. Although the tensile strength value of the prepared ceramic SiCN fibers is still lower, the continuous processing of thick SiCN fibers with diameter > 70 μm and tensile strength of ∼0.8 GPa is a significant result and a crucial precondition for their use as reinforcement in metal matrix composites.
 | ||
| Fig. 7 Effect of fiber diameter on the tensile strength for (a) ML33S or (b) HTTS derived ceramic SiCN fibers compared to glass fibers.24 | ||
In order to better understand the mechanical behavior of these ceramic SiCN fibers, Weibull distribution analysis was used to analyze the fracture strength. Considering that identical specimens of brittle materials, such as ceramic fibers, show a large variation of tensile fracture stresses, the cumulative distribution function proposed by Weibull25 was used for a statistical characterization of the tensile strength for ceramic SiCN fibers made from ML33S and HTTS. The single parameter Weibull distribution leads to a failure probability given by:
 | (02) |
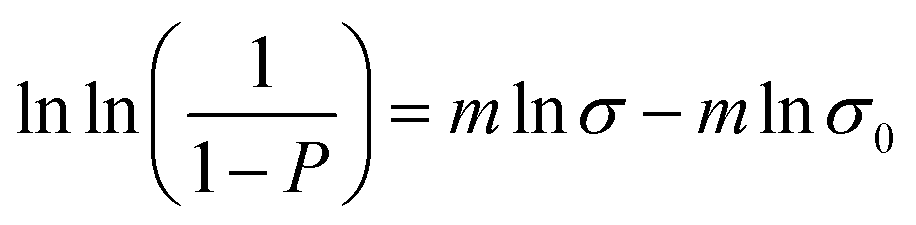 | (03) |
The Weibull analysis of brittle fracture is based on the weakest link theory, which assumes that all materials contain inhomogeneities, such as pores or flaws, distributed randomly with a certain density per unit volume. If these defects are the origin of material's fracture then tensile failure is determined by the largest critical defect.25,26
In Fig. 8, the results of the Weibull analysis on the strength of the ceramic SiCN fibers are presented. Although it is known that the variation in diameter influences the volume variation when the tensile strength is measured using the same gauge length (in this work being 25 mm) the single mode Weibull analysis was used and it was assumed that there is no volume variation as the fiber diameter changes.
 | ||
| Fig. 8 Weibull analysis of (a) ML33S and (b) HTTS derived ceramic SiCN fibers with diameter between 30 and 100 μm. | ||
The Weibull parameters, modulus m and scale parameter σ0 are presented in Table 2. The Weibull parameters are similar for the two types of fibers. In spite of the diameter variation from 30 to 100 μm, the single mode Weibull distribution fits the strength data reasonably well. The mean strength of ML33S and HTTS derived ceramic fibers is about 1 GPa.
| PDC | Fiber diameter (μm) |
![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) (μm) (μm) |
![[small sigma, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d2.gif) (MPa) (MPa) |
σ 0 (MPa) | m |
|---|---|---|---|---|---|
| ML33S | 30 < d < 100 | 65 ± 20 | 952 ± 254 | 1044 | 4.572 |
| HTTS | 30 < d < 100 | 61 ± 24 | 1001 ± 256 | 1122 | 4.462 |
For further analysis of the strength distribution, the strength results were grouped in two ranges – fiber diameter less than 70 μm and greater than 70 μm. Weibull analysis was conducted separately for these two groups and the results are shown in Fig. 9. As can be seen, the Weibull modulus of higher diameter fibers from ML33S and HTTS achieve values of m > 8, which implies a high reproducibility of the tensile strength of thick monofilaments. For thinner fibers, the Weibull modulus specially in the case of ML33S derived fibers is lower, being m = ∼6, reflecting the high scatter in the strength values for fibers with diameters of about 50 μm. In accordance with Griffith's observations and analysis, the average strength of the thin fibers was higher than that of thick fibers. The Weibull parameters for the thick and the thin ceramic fibers made from the two precursors are summarized in Table 3.
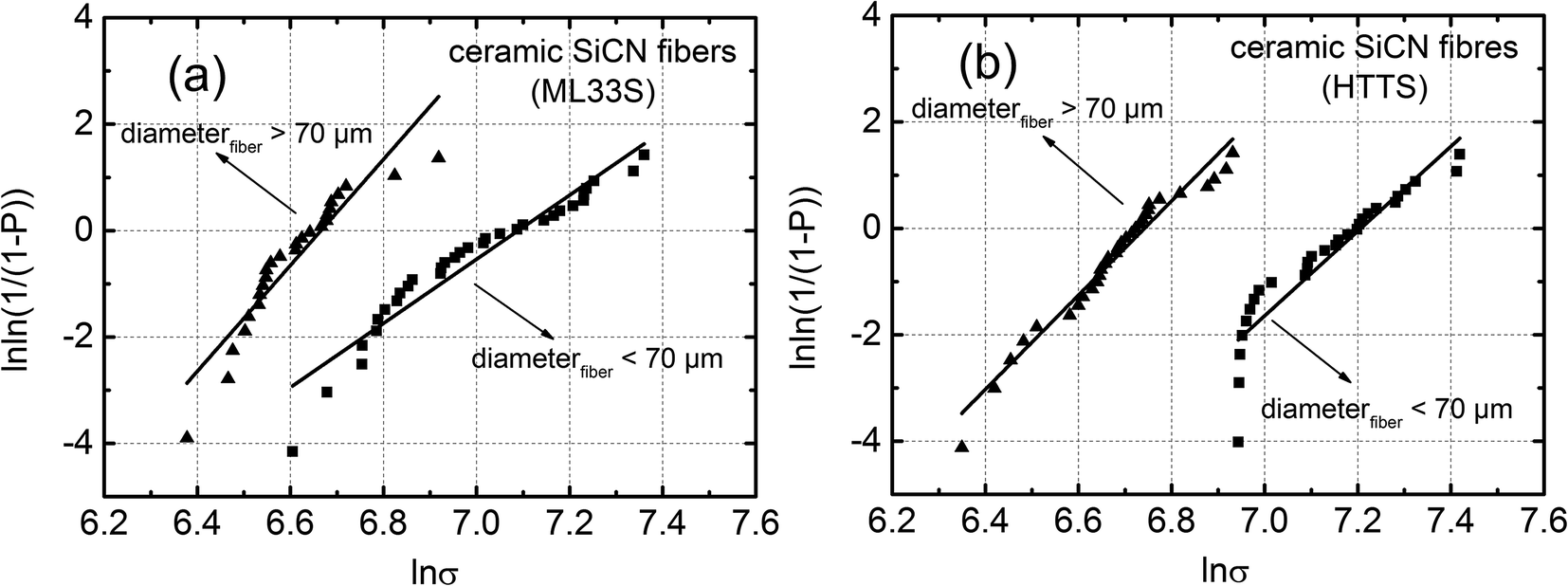 | ||
| Fig. 9 Weibull plots for (a) ML33S and (b) HTTS derived ceramic SiCN fibers divided in two groups – thin (diameter < 70 μm) and thick (diameter > 70 μm). | ||
| PDC | Fiber diameter (μm) |
(μm) |
(MPa) |
σ 0 (MPa) | m |
|---|---|---|---|---|---|
| ML33S | <70 | 49 ± 6 | 1113 ± 223 | 1200 | 6.022 |
| >70 | 85 ± 8 | 747 ± 91 | 785 | 9.957 | |
| HTTS | <70 | 36 ± 6 | 1268 ± 187 | 1348 | 7.957 |
| >70 | 81 ± 9 | 802 ± 110 | 847 | 8.855 |
As the strength of the SiCN ceramic fibers derived from ML33S or HTTS show similar values, it appears that the strength of the ceramic SiCN fibers is independent on the tensile strength of the cured green fibers. The results obtained in this work indicate that the strength of the uncured or cured green fibers is closely related to the chemical properties of the polymer, particularly the molecular weight, the rheology and the functional reactive groups. But in relation to ceramic SiCN fibers, the type of precursor, ML33S or HTTS, does not have significant influence on the strength.
A discussion focused on the dependency of the tensile strength of a polymer derived amorphous ceramic fiber on the fiber diameter, fiber surface and microstructure is rather complex. The strength distribution will depend on the polymer system, the processing history of the green fiber, their curing and pyrolysis conditions. Further investigations are needed including careful analysis of fracture origin in order to improve the strength and strength distribution of fibers made from precursors.
The creep behavior of polymer derived ceramic SiCN fibers, as well as SiC fibers, is correlated to their microstructure and mechanical stability. The factors, which will mainly influence the creep resistance of the ceramic fibers are processing temperature, oxygen and free carbon contents as already discussed in several publications.27–33
Fig. 10 demonstrates that both fiber types derived from ML33S and HTTS have similar creep resistance either in air as well as in nitrogen atmosphere. No stress relaxation occurs up to 1000 °C. Comparing the creep response of these fibers to other SiCN fibers, similar behavior is observed for ABSE polycarbosilazane derived SiCN fibers reported by Hacker,11 which were pyrolysed at 1300 °C containing 16 wt% of oxygen. When the test temperature for the ML33S and HTTS derived ceramic fibers is increased to 1100 °C the creep relaxation starts leading to decreased mBSR values. For creep test temperatures higher than 1100 °C, which exceed the processing temperature of the fibers, the mBSR values drops dramatically due to rearrangements within the amorphous SiCN phases.
 | ||
| Fig. 10 Bend stress relaxation modulus (mBSR) versus temperature for polymer derived ceramic fibers (dwell time at temperature: 1 hour). | ||
Similar to Nicalon SiC fibers,35 ML33S and HTTS derived ceramic fibers contain free carbon content up to ∼20 mol% (Table 4), high oxygen content (see Table 1) but they were pyrolysed at lower temperatures in comparison to Hi-Nicalon or Hi-Nicalon S fibers.37,38 However, Nicalon fibers already suffer stress relaxation at 1000 °C with the evolution of SiO and CO gases due to its microstructure, composed of an amorphous SiCO phase, a free carbon phase and small 2–5 nm β-SiC crystallites, compromising its creep resistance.35,36
| Microstructure | Stoichiometric formula (T = 1100 °C) | Free carbon (mol%) | |
|---|---|---|---|
| Ceramic from ML33S | Amorphous silicon (oxy)carbonitride + free carbon | SiC0.26N0.70O0.43 + 0.43Cf | 15.2 |
| Ceramic from HTTS | Amorphous silicon (oxy)carbonitride + free carbon | SiC0.29N0.23O1.07 + 0.67Cf | 20.6 |
In fact the creep resistance of SiCN fibers derived from ML33S and HTTS is similar to that of Hi-Nicalon SiC fibers, although they contain lower levels of oxygen and are processed at higher temperatures of ≥1300 °C. The better creep resistance of SiCN fibers in comparison to the oxygen rich Nicalon fibers is due to the stability of the amorphous SiCN phase against crystallization up to 1400 °C. An increase of the pyrolysis temperature for the processing of ML33S and HTTS derived fibers up to ∼1300 °C should also lead to a further increase of the creep resistance, as already demonstrated for ABSE derived ceramic SiCN fibers with about 16 wt% oxygen.11
As the focus of the present work was to develop large diameter SiCN monofilaments, it is instructive to compare the creep resistance of these fibers with other large diameter SiC fibers. Fig. 11 shows the creep resistance of large diameter (>70 μm) SiCN fibers from ML33S and HTTS in comparison to commercially available CVD SiC fibers. It can be noted that the mBSR modulus of the ceramic SiCN fibers prepared in this work is similar to the commercially available SCS-6 fibers and significantly better than that of Sigma fibers. The stress relaxation of Sigma fibers starts at 900 °C and the mBSR modulus is 0.8. At 1100 °C the creep resistance is very low and the BSR modulus is only ∼0.1.
 | ||
| Fig. 11 Bend stress relaxation modulus (mBSR) versus temperature for Si-based ceramic fibers with diameter > 70 μm (dwell time at temperature: 1 hour). | ||
Sigma 1156 fibers are composed of stoichiometric SiC grain near the tungsten core and excess silicon near the surface, which is responsible for the drop of the creep resistance at T > 900 °C.40 In contrast, the SiC in the SCS-6 fibers is nearly stoichiometric and chemically stable up to high temperatures. The creep behavior of these fibers in the temperature region 1100 < T < 1300 °C is dominated by microstructural changes in the complex composite structure of these fibers. SCS-6 fibers contain concentric regions with different SiC compositions, free carbon at the grain boundaries and a carbon-rich surface coating. This composite structure induces microstructural changes at high temperatures due to diffusion of free silicon from the near-stoichiometric region with β-SiC grains into the carbon-rich surface coating, reducing its thermal stability. Above 1300 °C the thermal degradation starts.39–41
The results in this chapter indicate that the more cost-effective precursor route for the processing of large diameter ML33S and HTTS derived ceramic SiCN fibers make their use as reinforcement of MMCs very attractive, despite their still limited mechanical properties. Although the maximum pyrolysis temperature of the polymer derived ceramic fibers prepared in this work is only 1100 °C, due to the limited maximum pyrolysis temperature of the furnace, their creep resistance is already comparable to SCS-6 fibers, which are currently the most preferred ceramic monofilaments to reinforce MMCs in spite of their high cost.
For the oxidation tests performed in this work, at temperatures between 900 and 1100 °C in air a thin and smooth passivating SiO2 coating forms on the surface of the fibers (CVD SCS-6, ML33S and HTTS derived ceramic fibers), protecting or reducing the oxidation of the material at these temperatures.
After annealing of the SCS-6 fibers in air at 1200 °C, an outer SiO2 layer with a thickness of 2.5 μm is formed (Fig. 12a). Cracks and pores within the oxide layer already appear after this oxidation test. Additionally, the carbon core is completely oxidized.
 | ||
| Fig. 12 SEM micrographs of the surface and cross section of ceramic fibers after 12 hours at 1200 °C in air: (a) CVD SCS-6 fiber, (b) ML33S and (c) HTTS derived ceramic SiCN fibers. | ||
In contrast, the oxide layer of the ML33S and HTTS derived fibers formed at the same conditions is smooth and defect free with a thickness between 1.5 and 1.8 μm (Fig. 12b and c and 13).
 | ||
| Fig. 13 Energy dispersive spectroscopy (EDS) (a) oxygen map and (b) silicon map of the cross section (SEM micrograph) of a (c) ML33S derived ceramic fiber after 12 hours at 1200 °C in air with (d) Si K and O K linescan. | ||
If ML33S or HTTS derived ceramic fibers are exposed to air at 1300 °C, the outer surface of the SiO2 layer with a thickness of ∼3.5 μm shows first signs of cracks (Fig. 14b and c). For comparison, the micrographs of the SCS-6 fiber also oxidized at 1300 °C are shown in Fig. 14a. The thickness of the resulting oxide layer is with about 3.5 μm comparable to our fibers.
 | ||
| Fig. 14 SEM micrographs of the surface and cross section of ceramic fibers after 12 hours at 1300 °C in air: (a) CVD SCS-6 fiber, (b) ML33S and (c) HTTS derived ceramic SiCN fibers. | ||
The oxidation behavior of the SiCN fibers most likely follows the same mechanisms as reported by Chollon and Mocaer10,43 and later on discussed by Hacker11 and Kokott.34 The ML33S and HTTS derived ceramic fibers can be considered as SiCN(O) systems, due to their oxygen contents >10 wt%. Therefore, the oxidation process in summary follows eqn (04):
| SiCxN4y/3O2(1−x−y)(s) + (3x/2 + y)O2(g) → SiO2(s) + xCO(g) + 2y/3N2(g) | (04) |
| C(s) + 1/2O2(g) → CO(g) | (05) |
During the oxidation of SiCN(O) systems a complex process occurs, involving the oxidation mechanism of both SiC and Si3N4, such as simultaneous evaporation of CO and N2 by diffusion through the oxide layer. According to Chollon,10 the high oxidation stability of SiCN(O) fibers is due to the formation of a SiCNO interface (composition SiC(x−δx)N4(y−δy)/3O2(1−x−y+δx+δy)) between the fiber core and the outer SiO2 layer. This interface enables a continuous variation of the concentration from initial SiCN(O) composition of the ceramic fiber until SiO2. Decisive for the oxidation stability is the necessary activation energy to further oxidize the SiCNO interface, which in this case is considered to be a combination of Si3N4, SiC and SiO2. The activation energy of the reaction of oxygen with Si3N4 is 330–490 kJ mol−1, which is much higher than with SiC, being 90–140 kJ mol−1. Therefore, the higher the content of Si–N bondings in the SiCN(O) system, the higher is the necessary activation energy for the oxidation reaction of the fiber and finally their oxidation stability.
Thus the oxidation tests indicate that the oxidation resistance of ML33S and HTTS derived ceramic SiCN fibers is higher or comparable to the commercially available SCS-6 fibers despite their low processing temperature of only 1100 °C. MMCs and CMCs are generally used to combine thermomechanical stability with oxidation resistance at high temperatures. If voids or cracks are present (or generated) in the matrix, the contact with oxygen degrades the reinforcement, depending on its oxidation resistance. Therefore, carbon fibers, thus also the carbon core of SCS-6 fibers (see Fig. 12a and 14a), are not suitable for high temperature applications in an oxidative atmosphere. In contrast ceramic SiCN fibers are completely protected with passivating oxide layers, which allows their use also in air at higher temperatures.
4. Conclusions
Ceramic SiCN fibers with diameters > 70 μm were successfully produced via melt-spinning of two recently developed solid polysilazanes from our chair, ML33S and HTTS, curing in inert atmosphere with electron beam irradiation doses up to 1000 kGy and continuous pyrolysis in inert atmosphere. After curing with an electron beam dose of 600 KGy, green fibers from ML33S achieve a tensile strength of 43 MPa and a gel content of 60%. Green fibers from HTTS cured with the same electron beam dose show higher strength values of 65 MPa and a gel content of 100%. This is a consequence of the more reactive Si-vinyl groups in HTTS, which assures a high level of cross-linking after electron beam treatment already with a low dose of only 200 kGy. The resulting cured green fibers are stronger and more flexible, which is very important for subsequent handling and pyrolysis to ceramic SiCN fibers.The tensile strength distribution of ceramic SiCN fibers is related to their fiber diameter. Ceramic SiCN fibers with diameters > 70 μm achieve a tensile strength of 800 MPa, while ceramic SiCN fibers with diameter < 70 μm yield strength values of ∼1.6 GPa. The Weibull modulus for the large diameter fibers is rather high (∼8) indicating that the strength variability is rather low.
The creep behavior of the ceramic SiCN fibers is influenced by their composition, mainly oxygen and free carbon contents, as well as by the pyrolysis temperature. No stress relaxation is observed for ML33S and HTTS derived ceramic SiCN fibers up to 1000 °C. Up to a temperature of 1100 °C, their creep behavior is similar to that of Nicalon and SCS-6 fibers. At higher temperatures the creep resistance decreases remarkably due to the low pyrolysis temperature of only 1100 °C and the high oxygen contents (12.9 and 28.6 wt%).
The oxidation resistance of ceramic SiCN fibers from ML33S and HTTS is similar or superior than SCS-6 fibers. After a heat treatment in air at 1200 °C for 12 hours, the surface of the ML33S and HTTS derived ceramic fibers is protected by a close defect free protective silica layer, while the oxidation layer of SCS-6 fibers already show cracks and pores.
For the first time we demonstrated the continuous processing of thick ceramic SiCN fibers by using the PDC technology. There is a high potential to improve the ceramic fiber properties by optimizing the processing parameters, reducing the oxygen content and increasing the pyrolysis temperature. Therefore the developed technology offers a promising route to produce thick ceramic fibers for e.g. the reinforcement of MMCs, especially titanium matrix composites.
5. Acknowledgments
The authors thank Ute Kuhn from the Polymer Engineering Institute and Andre Prette, University of Bayreuth, for the TGA and DSC measurements, and Fraunhofer Institute HTL, Bayreuth, for mechanical tests of the fibers. We also thank Deutsche Forschungsgemeinschaft (DFG) for supporting this work within the project SiMet. RKB acknowledges the Humboldt Research Award which was responsible for initiating this collaboration.References
- T. T. Cheng, I. P. Jones, R. A. Shatwell and P. Doorbar, Mater. Sci. Eng., A, 1999, 260, 139–145 CrossRef.
- O. Flores, R. K. Bordia, D. Nestler, W. Krenkel and G. Motz, Adv. Eng. Mater., 2014, 16, 621–636 CrossRef CAS.
- S. R. Nutt and F. E. Wawner, J. Mater. Sci., 1985, 20, 1953–1960 CrossRef CAS.
- A. R. Bunsell and A. Piant, J. Mater. Sci., 2006, 41, 823–839 CrossRef CAS.
- J. A. DiCarlo and H. M. Yun, Handbook of Ceramic Composites, Springer, New York, 2005, pp. 33–52 Search PubMed.
- G. Motz and R. K. Bordia, Handbook of textile fibre structure Volume 2: Natural, regenerated, inorganic and specialist fibres, Woodhead Publishing Ltd., Cambridge, UK, 2009, pp. 378–424 Search PubMed.
- M. Herkt, F. Heutling and U. Koch, Adv. Eng. Mater., 2004, 6, 761–767 CrossRef CAS.
- C. Leyens, J. Hausmann and J. Kumpfert, Adv. Eng. Mater., 2003, 5, 399–410 CrossRef CAS.
- P. Martineau, R. Pailler, M. Lahaye and R. Naslain, J. Mater. Sci., 1984, 19, 2749–2770 CrossRef CAS.
- G. Chollon, J. Eur. Ceram. Soc., 2000, 20, 1959–1974 CrossRef CAS.
- J. Hacker, Entwicklung einer preiswerten keramischen Faser für den Anwendungsbereich bis 1400 Grad Celsius auf der Basis eines spinnfähigen siliciumorganischen Polymers, Utz, München, 2006 Search PubMed.
- O. Flores, T. Schmalz, W. Krenkel, L. Heymann and G. Motz, J. Mater. Chem. A, 2013, 1, 15406–15415 CAS.
- O. Flores, L. Heymann and G. Motz, Rheo. Acta, 2015, 54, 517–528 CrossRef CAS.
- G. Motz, D. Decker and T. Schmalz, EP Patent, 12006145, 2012.
- DIN 16892 – Rohre aus vernetztem Polyethylen hoher Dichte (PE-X) Allgemeine Guteanforderungen, Prüfung, 6.5. Vernetzungsgrad, 2000.
- DIN EN 1007-4 – Hochleistungskeramik – Keramische Verbundwerkstoffe: Faserverstärkungen; Teil 4: Bestimmung der Zugeigenschaften von Fasern bei Raumtemperatur, 1994.
- G. N. Morscher and J. A. DiCarlo, J. Am. Ceram. Soc., 1992, 75, 136–140 CrossRef CAS.
- G. Motz, T. Schmalz, S. Trassl and R. Kempe, Design, Processing and Properties of Ceramic Materials from Preceramic Precursors, Nova Science Publishers Inc., Hauppauge USA, 2011, pp. 15–35 Search PubMed.
- T. Taki, K. Okamura, M. Sato, T. Seguchi and S. Kawanishi, J. Mater. Sci. Lett., 1988, 7, 209–211 CrossRef CAS.
- Z. Y. Chu, Y. C. Song, Y. S. Xu and Y. B. Fu, J. Mater. Sci. Lett., 1999, 18, 1793–1795 CrossRef CAS.
- A. Idesaki, M. Narisawa, K. Okamura, M. Sugimoto, Y. Morita, T. Seguchi and M. Itoh, Radiat. Phys. Chem., 2001, 60, 483–487 CrossRef CAS.
- S. Kokott and G. Motz, Soft Mater., 2007, 4, 165–174 CrossRef.
- D. Mocaer, R. Pailler, R. Naslain, C. Richard, J. P. Pillot, J. Dunogues, C. Darnez, M. Chambon and M. Lahaye, J. Mater. Sci., 1993, 28, 3049–3058 CrossRef CAS.
- A. A. Griffith, Philos. Trans. R. Soc. London, 1921, 221, 163–199 CrossRef.
- W. Weibull, J. Appl. Mech., 1951, 18, 293–297 Search PubMed.
- S. van der Zwaag, J. Test. Eval., 1989, 17, 292–298 CrossRef CAS.
- G. Chollon, R. Pailler, R. Naslain, F. Laanani, M. Monthioux and P. Olry, J. Mater. Sci., 1997, 32, 327–347 CrossRef CAS.
- G. Chollon, R. Pailler, R. Naslain and P. Olry, J. Mater. Sci., 1997, 32, 1133–1147 CrossRef CAS.
- P. le Coustumer, M. Monthioux and A. Oberlin, J. Eur. Ceram. Soc., 1993, 11, 95–103 CrossRef CAS.
- T. Mah, N. L. Hecht, D. E. McCullum, J. R. Hoenigman, H. M. Kim, A. P. Katz and H. A. Lipsitt, J. Mater. Sci., 1984, 19, 1191–1201 CrossRef CAS.
- G. Simon and A. R. Bunsell, J. Mater. Sci., 1984, 19, 3658–3670 CrossRef CAS.
- O. Delverdier, M. Monthioux, D. Mocaer and R. Pailler, J. Eur. Ceram. Soc., 1994, 14, 313–325 CrossRef CAS.
- M. Monthioux and O. Delverdier, J. Eur. Ceram. Soc., 1996, 16, 721–737 CrossRef CAS.
- S. Kokott-Wenderoth, Herstellung und Charakterisierung Multiwall-Carbon-Nanotube-verstärker keramischer SiCN-Fasern, Cuvillier Verlag, Göttingen, 2009 Search PubMed.
- R. E. Tressler, Composites, Part A, 1999, 30, 429–437 CrossRef.
- S. M. Johnson, R. D. Brittain, R. H. Lamoreaux and D. J. Rowcliffe, J. Am. Ceram. Soc., 1988, 71, C-132–C-135 CAS.
- G. N. Morscher and J. A. DiCarlo, Proceedings of the NASA 6th Annual HITEMP Review, Cleveland, OH, 1993 Search PubMed.
- M. Takeda, J. Sakamoto, A. Saeki and H. Ichikawa, Ceram. Eng. Sci. Proc., 1996, 17, 35–43 CAS.
- G. N. Morscher, C. A. Lewinsohn, C. E. Bakis and R. E. Tressler, J. Am. Ceram. Soc., 1995, 78, 3244–3252 CrossRef CAS.
- G. Chollon, R. Naslain, C. Prentice, R. Shatwell and P. May, J. Eur. Ceram. Soc., 2005, 25, 1929–1942 CrossRef CAS.
- R. T. Bhatt and D. R. Hull, J. Am. Ceram. Soc., 1998, 81, 957–964 CrossRef CAS.
- H. Du, R. E. Tressler, K. E. Spear and C. G. Pantano, J. Electrochem. Soc., 1989, 136, 1527–1536 CrossRef CAS.
- D. Mocaer, G. Chollon, R. Pailler, L. Filipuzzi and R. Naslain, J. Mater. Sci., 1993, 28, 3059–3068 CrossRef CAS.
Footnote |
| † On the occasion of Prof. Dr Gerhard Röwer's 75th birthday. |
| This journal is © The Royal Society of Chemistry 2015 |