
Direct hydrothermal reduction of graphene oxide based papers obtained from tape casting for supercapacitor applications†
Zheng Jia*a,
Chengyuan Lia,
Daoqing Liua and
Lixiang Jiangbc
aSchool of Chemical Engineering and Technology, Harbin Institute of Technology, Harbin 150001, China. E-mail: 13104059037@163.com
bScience and Technology on Reliability and Environmental Engineering Laboratory, Beijing 100094, China
cBeijing Institute of Spacecraft Environment Engineering, Beijing 100094, China
First published on 18th September 2015
Abstract
A novel method of producing flexible graphene-based paper (GBP) electrodes through the direct hydrothermal reduction of graphene oxide based paper is developed. On the basis of an electrostatic attraction mechanism or pH-enhanced π–π attraction mechanism, a delicate balance between integrity, flexibility and porosity of GBPs is maintained during the hydrothermal treatment process, providing them with excellent compositional, microstructural and supercapacitive characteristics. Furthermore, the insertion of CNT into the graphene interlayer spaces immensely improves the specific capacitance (up to 195 F g−1) and rate capability (10 A g−1) of the supercapacitors assembled with these GBPs. In addition, the facile and simple preparation procedures and arbitrarily controlled area and thickness of the GBPs predict their promising industrialization potential.
1. Introduction
Products from modern technology, like mobile phones and laptops, have made our daily life easier than ever, and to be fair, none of these would have happened without energy storage devices. With global awareness of increasingly severe environment and energy problems heightened, the appetite for these devices is growing stronger and stronger, and some devices are made to be flexible to meet the need of bendable and portable technological gadgets. Flexible energy storage devices require flexible electrodes, for which graphene is a promising candidate material.Up till now, researchers have developed several procedures to produce graphene-based papers (GBPs) that can be further made into flexible electrodes. The most commonly used is vacuum-assisted filtration either of graphene1,2 and reduced graphene oxide dispersion,3–6 or of graphene oxide (GO) dispersion followed by reduction.7–11 However, this procedure is time consuming, thus not economical enough to be put into massive production. Moreover, the areal energy densities of such GBP electrodes are restricted by their self-confined thickness, which is usually in several-micron range.
Another important procedure to fabricate GBP electrodes is the mechanical compression method.12–15 Some of them applied a freeze-drying method to produce GO-based aerogels, which were further converted into graphene-based aerogels by heat treatment, and then pressed into GBPs,12,13 while Yuxi Xu et al.14,15 mechanically compressed (holey) graphene hydrogels, which were obtained in hydrothermal processes, into GBP electrodes. In essence, this method compressed extremely porous graphene materials into integral films.
Other novel methods were also developed. Dong Zhang et al.16 applied positive and negative pulse electric signal to simultaneously form and reduce a GO thin paper, and tested its supercapacitive performances. Siyang Liu et al.17 obtained a GO paper from the liquid–air surface of heated GO dispersion, and reduced it with supercritical ethanol. Fei Xiao et al.18 casted a GO slurry onto a PTFE substrate and obtained a freestanding GO paper, and electrochemically reduced it to produce a flexible electrode.
The vital challenge of making GBPs for electrochemical energy storage devices is maintaining enough space between graphene layers for electrolyte access while keeping their integrity and flexibility at the same time. Actually, the affinity to keep the integrity and flexibility in the aforementioned cases originates from the localized crosslinking of graphene layers caused by π–π attraction, thus a delicate balance of the crosslinking degree should be kept so as to maintain the integrity and flexibility, and porosity as well.
In this study, we propose an electrostatic attraction mechanism for maintaining the integrity and flexibility of GBPs besides a pH-enhanced π–π attraction mechanism, and a novel procedure has been developed to fabricate GBPs on the basis of the proposed mechanisms. Unlike the GBPs fabricated by vacuum-assisted filtration, the thickness of these papers can be arbitrarily controlled by the adjustment of the concentration of the GO slurry and applied scale of the applicator. Therefore, the method we present here to fabricate binder-free, flexible GBPs is facile and simple and easy to obtain considerably large papers in area and thickness. Besides, since tape casting method and equipment are widely used in industry, and the conditions for hydrothermal reduction are easy to satisfy, this method is promising for commercialization.
2. Experimental
2.1 Preparation of GO dispersion
Graphite oxide was first prepared from natural graphite (Xinghe Co., Qingdao, China) using a modified Hummers method.19 Briefly, concentrated sulphuric acid, natural graphite, NaNO3 (Fengchuan Reagent, Tianjin, China) and KMnO4 (Fengchuan Reagent, Tianjin, China) powder were mixed sequentially together in an ice bath. Then the mixture was transferred to a water bath of 35 °C and 90 °C for further reaction. Then the as-prepared graphite oxide was washed with and dispersed into distilled water followed by ultrasonic exfoliation (Shumei KQ250DE, Kunshan, China) for an hour at 40 kHz to obtain GO dispersion.2.2 Preparation of GO and GO/CNT (carbon nanotube) papers
To prepare GO paper, the as-obtained GO dispersion was diluted to 1 mg mL−1 and further treated with sonication for 1 h to enhance its dispersibility. Then the GO dispersion was concentrated to about 10–20 mg mL−1 by a rotary evaporator (Yuhua Instrument Co., Gongyi, China) heated in a 70 °C water bath. The concentrated GO (cGO) dispersion saw an increase in viscosity, which was utterly crucial for its formation into a film, while still maintained its good dispersibility. Next, we used a Pushen KTO-55 adjustable film applicator (Shanghai Pushen Chemical Machinery Co. Ltd., China) to spread the cGO dispersion evenly onto a flat glass substrate. The glass was then placed horizontally under room temperature for the dispersion to dry. Once dried, we manually peeled off the as-obtained paper from the glass substrate.To prepare GO/CNT paper, similar procedures to those of fabricating GO paper were applied. Sequentially, GO dispersion was diluted and sonicated, followed by the addition of CNT (Timesnano, Chengdu, China) at a mass ratio of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (GO:CNT) and sonication for another 1 h to form a uniformly distributed GO/CNT composite dispersion. Then the composite dispersion was successively concentrated, spread onto a glass substrate with an applicator, horizontally placed and dried, and manually peeled off from the substrate. The detailed experimental parameters are the same as those of fabricating GO papers.
:1 (GO:CNT) and sonication for another 1 h to form a uniformly distributed GO/CNT composite dispersion. Then the composite dispersion was successively concentrated, spread onto a glass substrate with an applicator, horizontally placed and dried, and manually peeled off from the substrate. The detailed experimental parameters are the same as those of fabricating GO papers.
The thickness of GO and GO/CNT papers can be controlled by changing the scale of the applicator and the concentration of the corresponding slurry.
2.3 Preparation of rGO and rGO/CNT papers
The as-obtained GO and GO/CNT papers were hydrothermally reduced in aqueous solutions of 5 mol L−1 KOH and 5 mol L−1 H3PO4, respectively. The papers were first immersed into the solutions in hydrothermal reactors and hermetically sealed, and then 160 °C heated for 6 h. After heating, the reactor was cooled to room temperature and the reduced papers were taken out and washed till neutral. Finally, the papers were dried in an oven of 70 °C. The as-obtained reduced papers were labelled as P-K and P-P for rGO papers obtained from KOH and H3PO4 solutions, and P-KC and P-PC for rGO/CNT papers obtained from KOH and H3PO4 solutions, respectively.Additionally, we carried out an experiment where instead of acid and alkaline solutions, we used distilled water to immerse GO film. Similarly, it was then hydrothermally treated at 160 °C for 6 h and cooled to room temperature.
2.4 Characterization of rGO and rGO/CNT papers
X-ray diffraction (XRD) measurements were performed using a D/max–γB system (Cu Kα radiation, λ = 0.154178 nm, Rigaku, Japan). Scanning electron spectroscopy (SEM) was employed to study the morphology of the papers with a Quanta 200FEG (FEI, American). Energy-dispersive X-ray (EDX) elemental mapping was used to determine the elemental distribution on the GBP surfaces with an S-4800 (Hitachi, Japan). X-ray photoelectron spectroscopy (XPS) was measured to study the surface composition and chemical state with a K-Alpha system (Thermofisher Scientific Company) which used an Al Kα monochromator. Raman spectra were measured with a Renishaw inVia Raman spectrometer employing a 532 nm laser beam. The digital photos of the papers were taken with a Canon 650D.2.5 Assembly of symmetric supercapacitors with the rGO and rGO/CNT papers as electrodes and their electrochemical measurements
Each of the as-prepared paper was clipped into two self-supporting disk electrodes of the same weight, which were assembled into a symmetric supercapacitor in a CR2025-type cell container, with polypropylene membrane as the separator and 30 wt% KOH aqueous solution the electrolyte. The rGO or rGO/CNT loading densities of the electrodes were 1–2 mg cm−2. The four as-assembled coin-type cells were labelled as SCP-K, SCP-KC, SCP-P and SCP-PC, respectively, corresponding to the four different papers. Tests of cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were run with a CHI 660E electrochemical workstation (Chenhua, Shanghai, China). EIS tests were carried out at open circuit conditions using sinusoidal signals with an amplitude of 10 mV and a frequency range between 0.01 Hz and 100 kHz. The galvanostatic charge/discharge tests were carried out with a Neware CT-4008-5V50mA Battery Test System (Neware, Shenzhen, China). The CV and galvanostatic charge/discharge tests were performed in a voltage window of 1 V from 0 to 1 V, and test results were used to calculate specific capacitance (Cs) respectively usingwhere I is the current, and m is the mass of one electrode, and ΔV/t is the voltage variation rate.

3. Results and discussion
3.1 Morphology and microstructure
As is obviously shown in Fig. 1a, GO papers could be obtained through manually peeling off from glass substrate. The as-obtained GO paper shines a yellow color, which is typical to GO, and is flexible enough to be twisted and rolled. In addition, the size of the GO paper could be enlarged arbitrarily as long as the spreading area of GO slurry is large enough. The SEM image (Fig. 1b) shows that the GO paper surface is rather flat with some local exfoliation of graphene layers, suggesting that the paper is constituted of overlapped graphene layers. And the cross-sectional SEM image (Fig. 1c) further demonstrates that the graphene layers in the GO paper do parallel with each other in a face to face manner. The GO paper after hydrothermal reduction in distilled water is shown in Fig. 1d, where we can see that the GO paper experiences a complete destruction of its original form, indicating that the direct hydrothermal reduction of GO paper in distilled water is unfeasible. | ||
| Fig. 1 (a) Digital photos of a rolled and spreading (inset) GO paper. SEM images of the (b) surface and (c) cross section of the GO paper. (d) Digital photo of the GO paper after hydrothermal reduction in distilled water. | ||
We explain this phenomenon as follows. The overlapped electron clouds from the conjugated π bonds make adjacent graphene layers attract to each other, while the oxygen-containing functional groups on graphene layers carry negative charges after their dissociation in water, which make graphene layers electrostatically repel against each other. If the π–π attraction is greater than the electrostatic repulsion, the paper can retain its integrity. However, during the hydrothermal reduction in distilled water, graphene layers experience an immense increase in thermal movement, which greatly strengthens the repulsion and promotes the separation of graphene layers so that the graphene paper see a complete deformation.
The GBPs obtained from hydrothermal reduction in KOH and H3PO4 solutions are shown in Fig. 2. Unlike the GO paper after hydrothermal reduction in distilled water, P-K, P-P, P-KC and P-PC maintain their integrity and flexibility, and all give out a metallic luster that is typical to reduced graphene oxide. Additionally, the surfaces of P-K and P-KC are rougher, and seemingly thicker and fluffier than those of P-P and P-PC.
 | ||
| Fig. 2 Digital photos of (a) P-K, (b) P-P, (c) P-KC and (d) P-PC. Inset in each photo: photo of the corresponding bended GBP. | ||
We believe that the feasibility of direct hydrothermal reduction of GO-based papers in acidic or basic solutions without the decomposition of the papers is attributed to the enhancement of attraction effect of graphene layers in the papers. In acidic H3PO4 solutions, the negative charges of the oxygen-containing groups on graphene surfaces are partially shielded by their combination with H+ ions in the solutions, and thus the electrostatic repulsion between graphene layers is weakened. Accordingly, the π–π attraction becomes the dominant interaction between the adjacent graphene layers, thus the GO-based papers could stay intact regardless of the incremental thermal movement during the hydrothermal process. Such pH influences on the electrostatic interaction of GO have been demonstrated many times, such as in the studies by Dan Li et al.20 and Ruoff et al.21
As to the reduced papers obtained from the basic KOH solutions, graphene layers should be negatively charged more heavily, at the same time, K+ ions may intercalate into the voids of adjacent graphene layers (see the following details) and link the individual layers as a whole through an electrostatic attraction between positively charged K+ ions and negatively charged oxygen-containing groups, and thus maintain the integrity of the papers. Some researchers have reported that GO can self-assemble through interactions between oxygen-containing groups of GO and some metal ions with positive charges.22,23 Recently, we found that the electrostatic attraction between K+ ions and oxygen-containing groups of GO can exert an orientation and linkage effect to construct high-density graphene assembly materials with a regular, compact but microporous graphene packing structure.24 All these findings provide some kind of support for the hypothesis we propose here. Due to the intercalation effect of K+ ions, the as-obtained papers could enjoy an increased porosity between graphene layers, which is accountable for the fluffy appearance of the papers, and may facilitate the electrolyte accessibility and hence electrochemical performances as well.
Fig. 3a–d show the surface SEM images of the four papers, respectively. Compared with the flat surface of the GO paper (Fig. 1b), it can be seen that obviously corrugated structures appear on the surfaces of all of the papers after hydrothermal treatment, implying more pores are created in the papers caused by hydrothermal-promoted thermal movement. Besides, we can see that the addition of CNT significantly alters the surface morphology of the papers derived from both solutions. Especially the papers obtained from H3PO4 solution witness a more impressive morphological variation than their counterparts from KOH solution do, since tumor-shaped bumps are introduced onto the surface of P-PC (Fig. 3d). The reason is probably that the intercalated K+ ions expand the interlayer spaces and thus induce a more uniform distribution of CNTs in P-KC, alleviating the morphological change.
 | ||
| Fig. 3 SEM images of the surfaces of (a) P-K, (b) P-P, (c) P-KC and (d) P-PC. And cross-sectional SEM images of (e) P-K, (f) P-P, (g) P-KC and (h) P-PC. | ||
From the comparison of the cross-sectional SEM images (Fig. 3e–h), we can see that the papers derived from H3PO4 solution (P-P and P-PC) exhibit a denser layer stacking structure than that of their counterparts obtained from KOH solution (P-K and P-KC). This difference could be ascribed to the intercalation of K+ ions into interlayer spaces after hydrothermal treatment in KOH solution, which leads to the increase in interlayer spaces.
To further investigate the microstructure of the as-prepared papers, XRD tests were carried out and the results are shown in Fig. 4a. All the XRD patterns display a broad (002) diffraction peak centered at around 2θ = 23°, which corresponds to an interlayer spacing of 0.39 nm according to Bragg equation. And the relative intensity of the (002) diffraction peak signifies the order and compactness degree of graphene stacking. It can be seen from Fig. 4a that the peak intensity of P-P is almost twice as high as that of P-K, which indicates that the stacking compactness of graphene in P-K is poorer than that in P-P. In other words, P-K possesses a relatively more abundant porosity created by the intercalation of K+ ions than P-P. This is consistent with the observations from the cross-sectional SEM images in Fig. 3e and f. Furthermore, it's obvious that for the papers obtained from both KOH and H3PO4 solutions, the addition of CNT markedly reduces the intensity of the (002) diffraction peaks, which denotes an alleviated agglomeration of graphene layers and consequently enhanced porosity of paper as a whole due to the intercalation of CNTs between graphene layers. Considering that stacking of graphene layers in P-PC and P-KC is considerably disturbed, the marginal difference in peak intensity and diffraction angle is negligible.
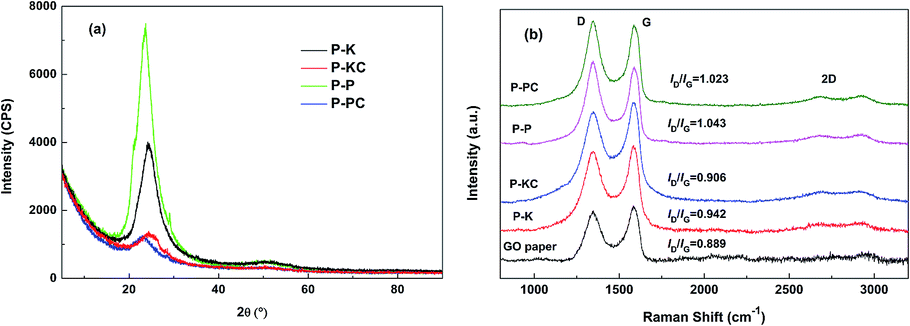 | ||
| Fig. 4 (a) XRD patterns of the as-prepared GBPs. (b) Raman spectra of the as-prepared GBPs and GO paper. | ||
Fig. 4b presents the Raman spectra of the as-prepared GBPs and GO paper. The intensity ratio (ID/IG) of the D band (at ∼1350 cm−1) and the G band (at ∼1590 cm−1) is widely used to quantify defects present in graphene-related materials.25 The results in Fig. 4b show that the ID/IG ratio increases from 0.889 for GO paper to 1.043 for P-P, which means more defects in P-P than in GO paper, probably due to the disintegration of graphene sheets into smaller sp2 graphene domains and the loss of carbon atoms caused by the decomposition of oxygen-containing functional groups.26 Compared with P-P, the ID/IG ratio of P-PC slightly rebounds to 1.023 due to the increase in sp2 domains caused by the addition of CNT. Comparatively, the ID/IG ratio is just lifted a little from 0.889 for GO paper to 0.942 for P-K, suggesting that relatively less defects were created after the hydrothermal reduction of GO paper in KOH medium, probably because the electrostatic linkage effect of K+ ions promotes the formation of bigger sp2 graphene domains, as we have ever observed.24 Similarly, the ID/IG ratio of P-KC slightly rebounds to 0.906 due to the increase in sp2 domains accompanied by the addition of CNT.
3.2 Elemental composition and surface chemistry
XPS tests were carried out to investigate the surface elemental composition and chemical state of the as-prepared GBPs. The survey XPS spectra (not shown) provide the surface elemental contents of the four papers, as listed in Table 1. It's clear that the oxygen contents of around 20 at% of the four papers indicate the partial reduction of GO after the hydrothermal treatment, and the papers obtained from KOH solution (P-K and P-KC) exhibit larger C/O ratios than their counterparts from H3PO4 solution (P-P and P-PC), probably meaning a little bit larger extent of reduction after hydrothermal treatment in KOH solution.| GBPs | C1s (at%) | O1s (at%) | C/O ratio | K2p (at%) | P2p (at%) | Relative contents of carbon bonds (%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (SP2) C (SP2) |
C–C (SP3) | C–O/C–O–C | CO |
O–CO |
||||||
| P-K | 76.63 | 23.37 | 3.28 | — | — | 65.92 | 10.74 | 16.02 | 4.73 | 2.59 |
| P-KC | 80.84 | 16.34 | 4.95 | 2.82 | — | 56.94 | 20.94 | 15.34 | 5.27 | 1.51 |
| P-P | 69.77 | 28.30 | 2.47 | — | 1.93 | 68.15 | 16.19 | 10.76 | 4.90 | |
| P-PC | 78.02 | 20.21 | 3.86 | — | 1.77 | 61.34 | 15.53 | 13.39 | 3.57 | 6.16 |
It also can be seen from Table 1 that phosphorus element is left in the papers after the hydrothermal process in H3PO4 solution (P-P and P-PC), while potassium element is just detected in P-KC (not P-K) by XPS technique. In order to determine whether potassium is present and how it is distributed in the papers prepared in KOH solution, the EDX potassium elemental mappings on the surfaces of P-K and P-KC were measured, as shown in Fig. 5.
 | ||
| Fig. 5 SEM images (a and c) and the corresponding EDX K elemental mappings (b and d) of the surfaces of P-K (a and b) and P-KC (c and d). | ||
It can be seen from Fig. 5b and d that K element definitely intercalates in and distributes rather uniformly across both P-K and P-KC although the K amount in P-K is markedly less than in P-KC. The difference of K amount in these two papers may be related to the richer pores created by the insertion of CNTs in P-KC. Perhaps, it is these K+ ions intercalated into the graphene interlayer spaces that maintains the integration of these two papers through the electrostatic attraction between the positively charged K+ ions and the neighboring negatively charged graphene layers.
Fig. 6 presents the C1s XPS spectra of the four papers, and the decomposed peaks representing various carbon bonds are summarized into Table 1. It can be shown that the relative contents of the C–O/C–O–C single bond component and CO double bond component in the papers obtained in KOH solution (P-K and P-KC) are higher than those in the papers obtained in H3PO4 solution (P-P and P-PC), while P-P and P-PC are richer in O–CO double bond component. Besides, it is worthwhile to point out that two peaks corresponding to K2p are determined in P-KC, confirming the presence of K element.
 | ||
| Fig. 6 C1s XPS spectra of (a) P-K, (b) P-KC, (c) P-P and (d) P-PC. | ||
3.3 Electrochemical performances of the GBP-based symmetric supercapacitors
Fig. 7 gives the specific capacitance plots transformed from the CV curves of SCP-K, SCP-P, SCP-KC and SCP-PC. Basically, all of the plots of the four supercapacitors exhibit quasi-rectangular shape at low scan rates, signifying an electric-double-layer (EDL) capacitive behavior. However, their different current change speeds at the transition point of the scan direction disclose their different capacitive characteristics. Actually, this index primarily depends on the RC time constant, and hence is closely related to the resistance of the system. Although the reduction degree of P-K is higher than that of P-P (as implied by higher C/O ratio of P-K in Table 1), the capacitive characteristic of SCP-K is poorer than that of SCP-P, probably due to the higher resistance of P-K. We speculate that the reason is that the electronic conduction between graphene layers in P-K is worse than P-P because the linkage of graphene layers in P-K is realized mainly through the mediation of K+ ions, not the direct overlapping of electron clouds from the conjugated π bonds of adjacent graphene layers, as in the case of P-P. Nevertheless, it is obvious that the addition of CNT profoundly improves the capacitive characteristics and rate capability of the papers obtained from the both aqueous solutions. Especially for SCP-KC, the quasi-rectangular curve shape is even retained at scan rates as high as 500 mV s−1, and the capacitance decay with scan rate is the lowest among the four capacitors. The reason may be that the insertion of highly conductive CNTs provides a new pathway for electronic conduction between graphene layers, which alleviates the conductivity problem, and more interlayer pores are created by the inserted K+ ions and CNTs, which facilitate the penetration of the electrolyte into the paper to fabricate more interface electric double layers. Another fact that should be pointed out is that the varied capacitance values of these curves from the low voltage range to high voltage range reveal the existence of pseudo-capacitance provided by Faradaic redox reactions of oxygen-containing groups besides the EDL capacitance.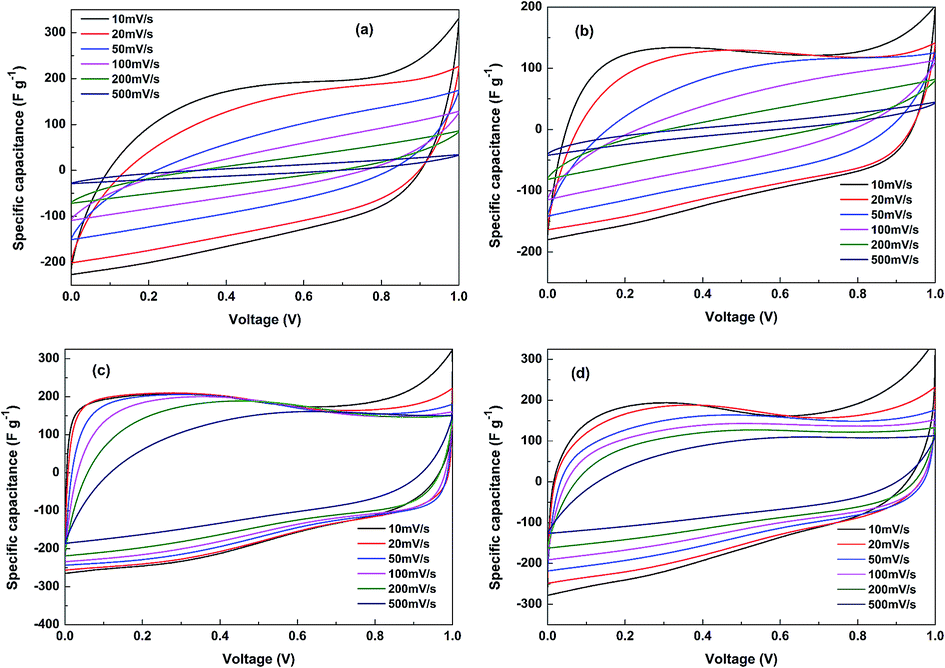 | ||
| Fig. 7 Cyclic voltammograms of (a) SCP-K, (b) SCP-P, (c) SCP-KC and (d) SCP-PC. | ||
It can be seen from Fig. 8 that at 0.2 A g−1 the discharge specific capacitance of SCP-K (191 F g−1) is markedly larger than that of SCP-P (144 F g−1), plausibly ascribed to more pores created by the inserted K+ ions in P-K. However, worse rate capability is witnessed for SCP-K, whose specific capacitance almost vanishes at 10 A g−1, probably due to the lower electronic conductivity of P-K. Comparatively, the addition of CNT significantly enhances the electrochemical performances. For SCP-KC, the discharge specific capacitance just decreases from 195 F g−1 at 0.2 A g−1 to 127 F g−1 at 10 A g−1, and for SCP-PC, the discharge specific capacitance is 174 F g−1 at 0.2 A g−1 and 91 F g−1 at 10 A g−1. Such huge improvements should be related to the ameliorated electronic conductivity and porosity caused by the insertion of CNT. And these tendencies are all consistent with the CV results in Fig. 7.
 | ||
| Fig. 8 Galvanostatic charge/discharge capacitance values of (a) SCP-K, (b) SCP-P, (c) SCP-KC and (d) SCP-PC at different current densities. | ||
In order to verify the proposed interpretation above for the electrochemical measurement results, the equivalent series resistance (ESR, in mΩ g) values are derived from the initial nearly vertical voltage drop at the transition point from charge to discharge (within 0.1 s) for all the four capacitors at different current densities, as presented in Fig. 9. The ESR values are calculated according to,
| ESR = ΔV/ΔI |
 | ||
| Fig. 9 Equivalent series resistance (ESR) values of (a) SCP-K, (b) SCP-P, (c) SCP-KC and (d) SCP-PC at different current densities. | ||
Under small current densities such as 0.2, 0.5 and 1 A g−1, the initial vertical voltage drop is too small to be measured precisely, and thus the ESR values derived at these current densities are not reliable. Therefore, only the ESR values obtained from data at current densities larger than 1 A g−1 are averaged to give the final ESR values; and for SCP-K, due to its overlarge ESR value, when the current density surpasses 5 A g−1, the initial vertical voltage drop values determined from the curve practically approach the testing voltage range (1 V) and are actually lower than their real values so that the derived ESR values at 8 and 10 A g−1 are underestimated. Hence, the nearly unvaried ESR values obtained at 1.5, 2, 3 and 5 A g−1 are averaged to give the final ESR value of SCP-K.
As we can see from Fig. 9, SCP-K exhibits the biggest ESR, which is 60.4 mΩ g, and followed by SCP-P, SCP-PC and SCP-KC with ESR values of 27.9, 9.8 and 8.3 mΩ g, respectively. Theoretically, the ESR reflects the ohmic resistance of a superapacitor if the measurement time is short enough (i.e., the measured initial voltage drop is only the ohmic voltage drop), which is composed of the electronic resistance of the electrodes (the GBP electrodes in this case) and ionic resistance of the electrolyte through the GBP electrodes and separator. Because the ionic conductivity of an aqueous supercapacitor electrolyte is rather high and the same electrolyte is used in these four GBP-based supercapacitors, the difference in the ESR value of them is primarily related to the different electronic resistances of these GBPs.
As we have mentioned above, the electronic conductivity of P-K should be poorer than P-P because of the indirect electronic conduction across graphene layers in P-K through the mediation of K+ ions, not the direct overlapping of electron clouds from the conjugated π bonds of adjacent graphene layers, as in the case of P-P. Therefore, the ESR value of SCP-K is greatly larger than that of SCP-P. When CNTs are inserted into the interlayer spaces of these two papers, the electronic conductivity is significantly enhanced due to the new electronic conduction networks provided by the conductive CNTs. Accordingly, the ESR values of SCP-KC and SCP-PC are markedly reduced in comparison with their counterparts without CNTs.
Because the initial voltage drop is actually measured within the first 0.1 s (not in an infinitely short period) after the current switches from charge to discharge, an electrolyte diffusion impedance across the papers is included in the ESR values besides the ohmic resistance. And the intercalation of K+ ions in P-KC creates more interlayer pores compared to P-PC, which facilitates the reduction of the electrolyte diffusion impedance, thus the ESR value of SCP-KC is a little bit smaller than that of SCP-PC. Consequently, the electrochemical performances (specific capacitance and rate capability) of SCP-KC are superior to those of SCP-PC, as shown in Fig. 7 and 8.
From the EIS plots in Fig. 10, we can see that the sizes of the high-frequency capacitive arcs of the four supercapacitors, which represent the impedance at a frequency around several Hz (equivalent to a discharge time of around 0.1–1 s), display an order exactly the same as that of the ESR values, as shown in Fig. 9. The capacitive arc size of SCP-K is the biggest, followed by SCP-P, SCP-PC and SCP-KC in a decreasing sequence. As discussed above, this order is attributed to the microstructure characteristics of the four GBPs, and coincides with their electrochemical performances, as shown in Fig. 7 and 8.
 | ||
| Fig. 10 Nyquist plots of the symmetric capacitors assembled with the as-prepared GBPs. | ||
Additionally, the cycling stability of SCP-KC and SCP-PC are measured, as shown in Fig. 11. The initial specific capacitance of SCP-KC is 171 F g−1 at 1 A g−1, and after 1000 cycles 167 F g−1 is retained. For SCP-PC, specific capacitance starts from 161 F g−1 and reaches a plateau of 167 F g−1 for about 250 cycles from around the 100th cycle, and eventually ends at 161 F g−1. It is obvious that after 1000 cycles of charge/discharge, the specific capacitance barely changes, which indicates excellent cycling stability. The columbic efficiency of both SCP-KC and SCP-PC rarely changes during the 1000 cycles, approaching 100%.
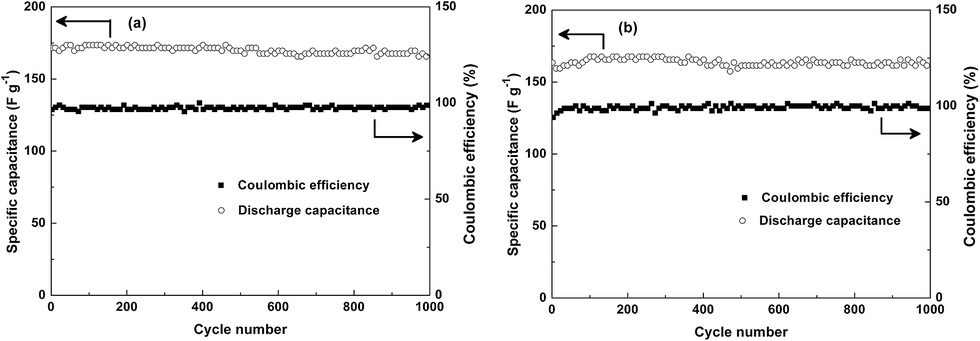 | ||
| Fig. 11 Cycling performance curves of (a) SCP-KC and (b) SCP-PC at 1 A g−1 current density. | ||
Conclusions
In summary, it is proved feasible to directly reduce GO based papers hydrothermally in an acidic or basic solution. The as-obtained GBPs can not only maintain their integrity and flexibility, but also create abundant graphene interlayer pore spaces available for the electrolyte access to form interface electric double layers. The systematic comparison of the GBPs obtained from H3PO4 and KOH solutions reveals the similarities and differences of their compositional, microstructural and supercapacitive characteristics, and discloses the intrinsic relationship of these characteristics. Last but not least, the proposed electrostatic attraction mechanism, pH-enhanced π–π attraction mechanism and those consequential properties of the as-obtained graphene assembly materials provide us insight into the nature of graphene packing structure, which might be enlightening for further investigations.Acknowledgements
This study was supported by the opening fund of Science and Technology on Reliability and Environmental Engineering Laboratory, China.Notes and references
- H. Bi, J. Chen, W. Zhao, S. Sun, Y. Tang, T. Lin, F. Huang, X. Zhou, X. Xie and M. Jiang, RSC Adv., 2013, 3, 8454–8460 RSC.
- G. Ning, C. Xu, Y. Cao, X. Zhu, Z. Jiang, Z. Fan, W. Qian, F. Wei and J. Gao, J. Mater. Chem. A, 2013, 1, 408–414 CAS.
- H. Gwon, H.-S. Kim, K. U. Lee, D.-H. Seo, Y. C. Park, Y.-S. Lee, B. T. Ahn and K. Kang, Energy Environ. Sci., 2011, 4, 1277–1283 CAS.
- L. Tang, Y. Wang, Y. Li, H. Feng, J. Lu and J. Li, Adv. Funct. Mater., 2009, 19, 2782–2789 CrossRef CAS PubMed.
- Y. Sun, Z. Fang, C. Wang, K. R. R. M. Ariyawansha, A. Zhou and H. Duan, Nanoscale, 2015, 7, 7790–7801 RSC.
- Z. Xu, H. Gao and H. Guoxin, Carbon, 2011, 49, 4731–4738 CrossRef CAS PubMed.
- M. Kim, D. Y. Kim, Y. Kang and O. O. Park, RSC Adv., 2015, 5, 3299–3305 RSC.
- C. Li, Y. Hu, M. Yu, Z. Wang, W. Zhao, P. Liu, Y. Tong and X. Lu, RSC Adv., 2014, 4, 51878–51883 RSC.
- T. Rath and P. P. Kundu, RSC Adv., 2015, 5, 26666–26674 RSC.
- L. L. Zhang, X. Zhao, M. D. Stoller, Y. Zhu, H. Ji, S. Murali, Y. Wu, S. Perales, B. Clevenger and R. S. Ruoff, Nano Lett., 2012, 12, 1806–1812 CrossRef CAS PubMed.
- J. Zhu, L. Zhu, Z. Lu, L. Gu, S. Cao and X. Cao, J. Phys. Chem. C, 2012, 116, 23075–23082 CAS.
- C. Wang, X. Wang, Y. Wang, J. Chen, H. Zhou and Y. Huang, Nano Energy, 2015, 11, 678–686 CrossRef CAS PubMed.
- F. Liu, S. Song, D. Xue and H. Zhang, Adv. Mater., 2012, 24, 1089–1094 CrossRef CAS PubMed.
- Y. Xu, Z. Lin, X. Huang, Y. Liu, Y. Huang and X. Duan, ACS Nano, 2013, 7, 4042–4049 CrossRef CAS PubMed.
- Y. Xu, Z. Lin, X. Zhong, X. Huang, N. O. Weiss, Y. Huang and X. Duan, Nat. Commun., 2014, 5, 4554 CAS.
- Y. Liu, D. Zhang, Y. Shang and Y. Liu, RSC Adv., 2014, 4, 30422–30429 RSC.
- S. Liu, K. Chen, Y. Fu, S. Yu and Z. Bao, Appl. Surf. Sci., 2012, 258, 5299–5303 CrossRef CAS PubMed.
- F. Xiao, J. Song, H. Gao, X. Zan, R. Xu and H. Duan, ACS Nano, 2012, 6, 100–110 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- D. Li, M. B. Mueller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- H. Bi, K. Yin, X. Xie, Y. Zhou, N. Wan, F. Xu, F. Banhart, L. Sun and R. S. Ruoff, Adv. Mater., 2012, 24, 5124–5129 CrossRef CAS PubMed.
- S. Park, K.-S. Lee, G. Bozoklu, W. Cai, S. T. Nguyen and R. S. Ruoff, ACS Nano, 2008, 2, 572–578 CrossRef CAS PubMed.
- S. Salgado, L. Pu and V. Maheshwari, J. Phys. Chem. C, 2012, 116, 12124–12130 CAS.
- D. Liu, Z. Jia, J. Zhu and D. Wang, J. Mater. Chem. A, 2015, 3, 12653–12662 CAS.
- X. Gao and X. Tang, Carbon, 2014, 76, 133–140 CrossRef CAS PubMed.
- J. Yan, Q. Wang, T. Wei, L. L. Jiang, M. L. Zhang, X. Y. Jing and Z. J. Fan, ACS Nano, 2014, 8, 4720–4729 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra17277b |
| This journal is © The Royal Society of Chemistry 2015 |