
On the morphology of MoS2 slabs on MoS2/Al2O3 catalysts: the influence of Mo loading†
Abstract
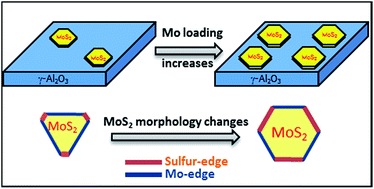
Two-dimensional MoS2 is an important material with diverse catalytic applications. The edge sites exposed by MoS2 are of great importance to its catalytic performance since the catalytic reactions generally occur on the edge sites rather than on the basal planes. In this work, low temperature (100 K) CO adsorption followed by IR spectroscopy (IR/CO) was used to in situ probe the edge sites of the MoS2 phase on MoS2/Al2O3 catalysts. It is found that the morphology of the MoS2 phase on an Al2O3 support is a truncated triangle exposing mainly Mo-terminated edges (M-edge). However, the proportion of sulfur-terminated edges (S-edge) increases with increasing the Mo loading, resulting in a more heavily truncated triangle morphology of MoS2. The change of MoS2 morphology with Mo loading can be explained by the modification of MoS2–Al2O3 interactions, indicating the importance of phase-support interactions in supported MoS2 catalysts.
 Please wait while we load your content...
Please wait while we load your content...

