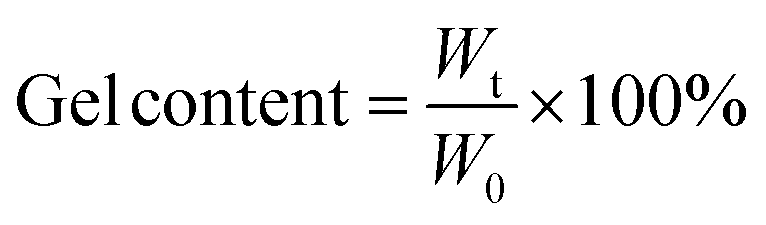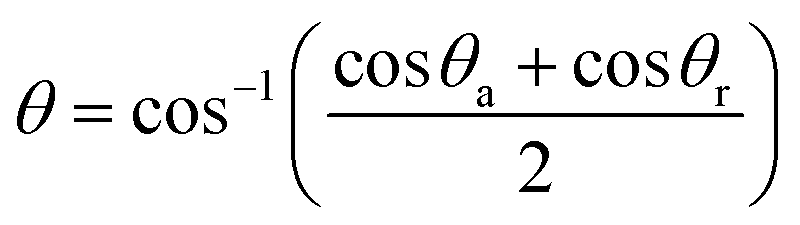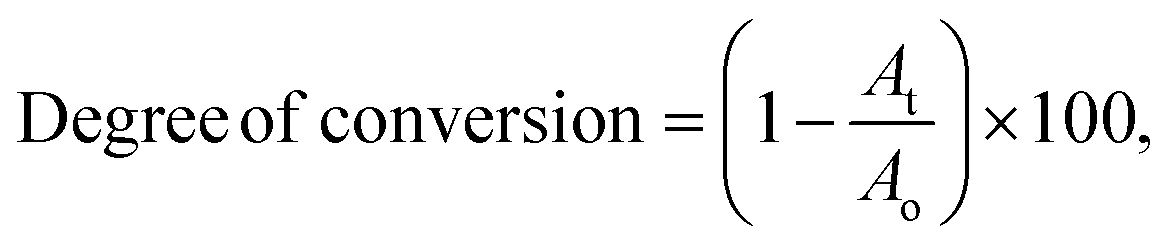DOI:
10.1039/C5RA17240C
(Paper)
RSC Adv., 2015,
5, 81838-81846
Synthesis and characterization of UV-curable acrylate films modified by functional methacrylate terminated polysiloxane hybrid oligomers
Received
26th August 2015
, Accepted 11th September 2015
First published on 11th September 2015
Abstract
A series of novel methacrylate terminated polysiloxane hybrid oligomers and functional acrylate oligomers were synthesized and characterized by GPC, FT-IR and NMR. The functional polysiloxane oligomers were introduced into the acrylate UV-curing system to improve its surface and thermal properties. With increasing the organosiloxane content, the contact angles of the UV-cured films increased, suggesting that the organosiloxane segments migrated to the top surface. The SEM and EDS results demonstrated the migration of the organosiloxane segments. The refractive index results showed that the optical performance did not decrease after the organosiloxane segments were incorporated. According the TGA curves, the decomposition temperatures of the polysiloxane/acrylate composite UV-cured films were higher than that of the pure acrylate UV-cured film, which demonstrated that the organosiloxane groups enhanced the thermal properties of the acrylate film due to the high energy of the Si–C bond. The observation of the fractured-surface morphology showed that the organosiloxane segments floated on the surface of the UV-cured films.
1. Introduction
UV-curing polymerization is widely used because of its beneficial properties1–4 such as rapid curing speed, clean and efficient energy, VOC free, and moderate curing conditions.5–8 (Meth)acrylate groups have been employed widely for photopolymerization because of their strong reactivity, their optical clarity characteristics, mechanical properties, adhesion and chemical stability, and showing rapid, near-complete conversions (i.e. on residual unreacted monomers) with low heat generation.9 Through the proper selection of acrylic/methacrylic monomers and curing agents, the cured polymers can be tailored to specific performance characteristics. Acrylic/methacrylic polymers form materials that are well known for their uses and applications in many important fields, especially in the formulation of paints and surface coatings.10,11 In these applications however, materials with poor hydrophobicity are not useful unless they are modified. The acrylic/methacrylic polymers are inferior to some silicon-containing materials in terms of elasticity, flexibility, hydrophobicity and heat resistance.12,13 To meet the need for high-performance coatings in high-technology areas, the UV-curable acrylic/methacrylic coating formulations must be continuously improved.
Polysiloxanes are the most important class of polymers with a non-carbon backbone, exhibiting a large degree of main-chain flexibility and high thermal stability due to Si–O groups,14–18 therefore, polysiloxanes have attracted much interest and are widely used in modifying polyacrylate coatings. However, due to differences in solubility parameters polysiloxanes are not very compatible with polyacrylates. To solve this problem, many efforts have been made for the purpose of combining these two materials through chemical methods. Bourgeat-Lami, Bai and Zhao et al. researched the synthesis of polysiloxanes and polyacrylates through emulsion polymerization.19–21 Yu et al. synthesized and evaluated two series of polyacrylate–polydimethylsiloxane (PDMS) block and graft copolymers used in anti-icing coatings.22 Moreover, research is focused on incorporating polysiloxanes into the main chain of a polymer, most frequently by emulsion polymerization methods, to improve the compatibility. However, emulsion polymerization products show poor hydrophobicity. Thus, there is an ever increasing demand for polysiloxane modified polyacrylates with better defined, improved and novel physical, chemical and mechanical properties. In this study, we prepared polysiloxane oligomers and then introduced acrylic double bonds by the hydrolysis reaction with methacryloxy propyl trimethoxysilane at a certain stage. Methacrylate terminated polysiloxane (MATSi) was thus obtained. Owing to the partly similar structure of methacryloxy propyl trimethoxysilane and polyacrylate, the compatibility between the functional polysiloxane and polyacrylate could be improved.
The polyacrylate in this study was synthesized as a novel UV-curable polyacrylate (PA). 6-Methylheptyl methacrylate was used as the major monomer because it provided good plasticity and was economical. Minor amounts of hydroxyethylmethacrylate were added to promote adherence properties. Glycidyl methacrylate was used to introduce epoxy groups to react with methacrylate (MA) in the following reaction step. Furthermore, a series of organosiloxane modified polyacrylates (OSPAs) at different organosilicon concentration ratios were respectively prepared by mixing PA with MATSi. In the presence of a photoinitiator, OSPAs were cross-linked by the radical polymerization of the carbon–carbon double bonds using UV irradiation and then the OSPA cured composite coatings were quickly prepared. In this way, various properties and enhanced performance could be obtained due to the OSPA cross-linked structure. The obtained polymers were characterized by gel permeation chromatography (GPC), Fourier transform infrared (FT-IR) spectroscopy and nuclear magnetic resonance (NMR). Furthermore, the surface hydrophobic, optical, and thermal properties of the cross-linked coatings made from the obtained polymers were investigated by the gel content test, flexibility test, pencil hardness test, contact angle (CA) analysis, refractive index test, scanning electron microscopy, energy dispersive spectrometry, differential scanning calorimetry thermograms and thermogravimetric analysis.
2. Experimental
2.1 Materials
Octaphenylpolyoxyethylene (OPE) was obtained from Quzhoumingfeng chemical company. Irgacure 1173 was obtained from Ciba Specialty Chemicals. Dodecylbenzene sulfonic acid (DBSA), glycidyl methacrylate (GMA), hydroxyethylmethacrylate (HEMA), 6-methylheptyl methacrylate (MHMA), methacrylic acid (MA) and methacryloxy propyl trimethoxysilane (KH570) were purchased from Aladdin Industrial Corporation. Octamethylcyclotetrasiloxane (D4) was provided by Guangzhou Jiahua chemical company. Distilled water, methanol, triethanolamine and azobisisobutyronitrile (AIBN) were purchased from Jingke Chemical Glass Instrument Co., Ltd. Butanone, tetrabutyl ammonium bromide (TBAB) and ethylalcohol were purchased from Sinopharm Chemical Reagent Co., Ltd. All the above materials were used as received without further purification.
2.2 Synthesis of polyacrylate
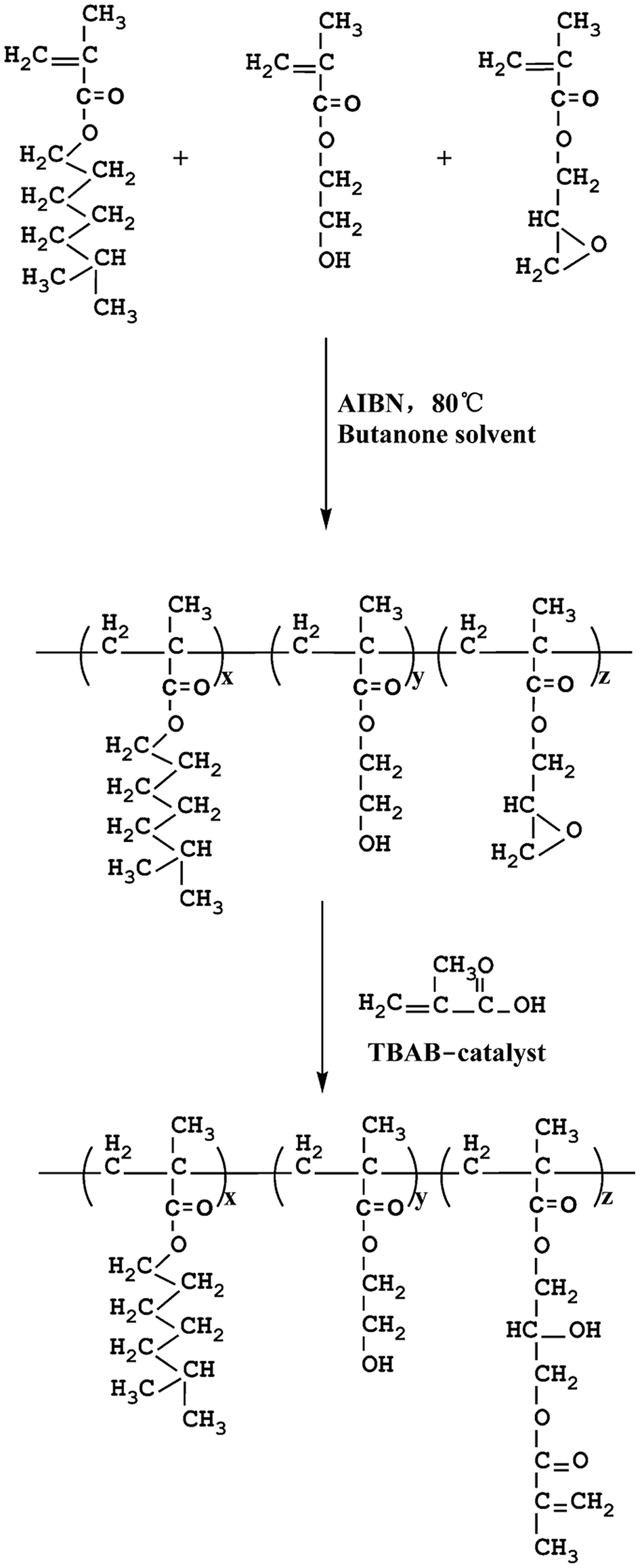
The polyacrylate was prepared using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:8 weight ratio of GMA, HEMA and MHMA in the presence of 4 wt% AIBN as initiator and butanone as solvent at a temperature of 80 °C. The reaction was carried out in a four-neck reaction kettle equipped with mechanical stirrer, nitrogen inlet, water condenser and thermometer. The reaction was carried out for 8 h to obtain the desired product. Subsequently, butanone was removed by reduced pressure distillation. The retained desired product was labeled as GHM and was transported into another four-neck kettle. Afterwards, a certain amount of MA was added into the kettle in the presence of 0.8 wt% TBAB. The reaction was carried out at 102 °C to reach 99.5% conversion, determined by standard acid value. After the remaining butanone and unreacted MA were discarded by reduced pressure distillation, the product was obtained and labeled as PA. The general procedure is shown in Scheme 1.
:1:8 weight ratio of GMA, HEMA and MHMA in the presence of 4 wt% AIBN as initiator and butanone as solvent at a temperature of 80 °C. The reaction was carried out in a four-neck reaction kettle equipped with mechanical stirrer, nitrogen inlet, water condenser and thermometer. The reaction was carried out for 8 h to obtain the desired product. Subsequently, butanone was removed by reduced pressure distillation. The retained desired product was labeled as GHM and was transported into another four-neck kettle. Afterwards, a certain amount of MA was added into the kettle in the presence of 0.8 wt% TBAB. The reaction was carried out at 102 °C to reach 99.5% conversion, determined by standard acid value. After the remaining butanone and unreacted MA were discarded by reduced pressure distillation, the product was obtained and labeled as PA. The general procedure is shown in Scheme 1.
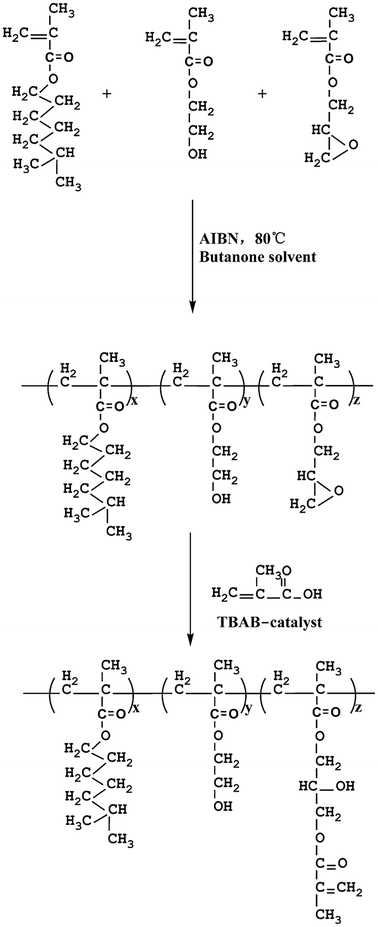 |
| | Scheme 1 Synthesis route of UV-cured PA. | |
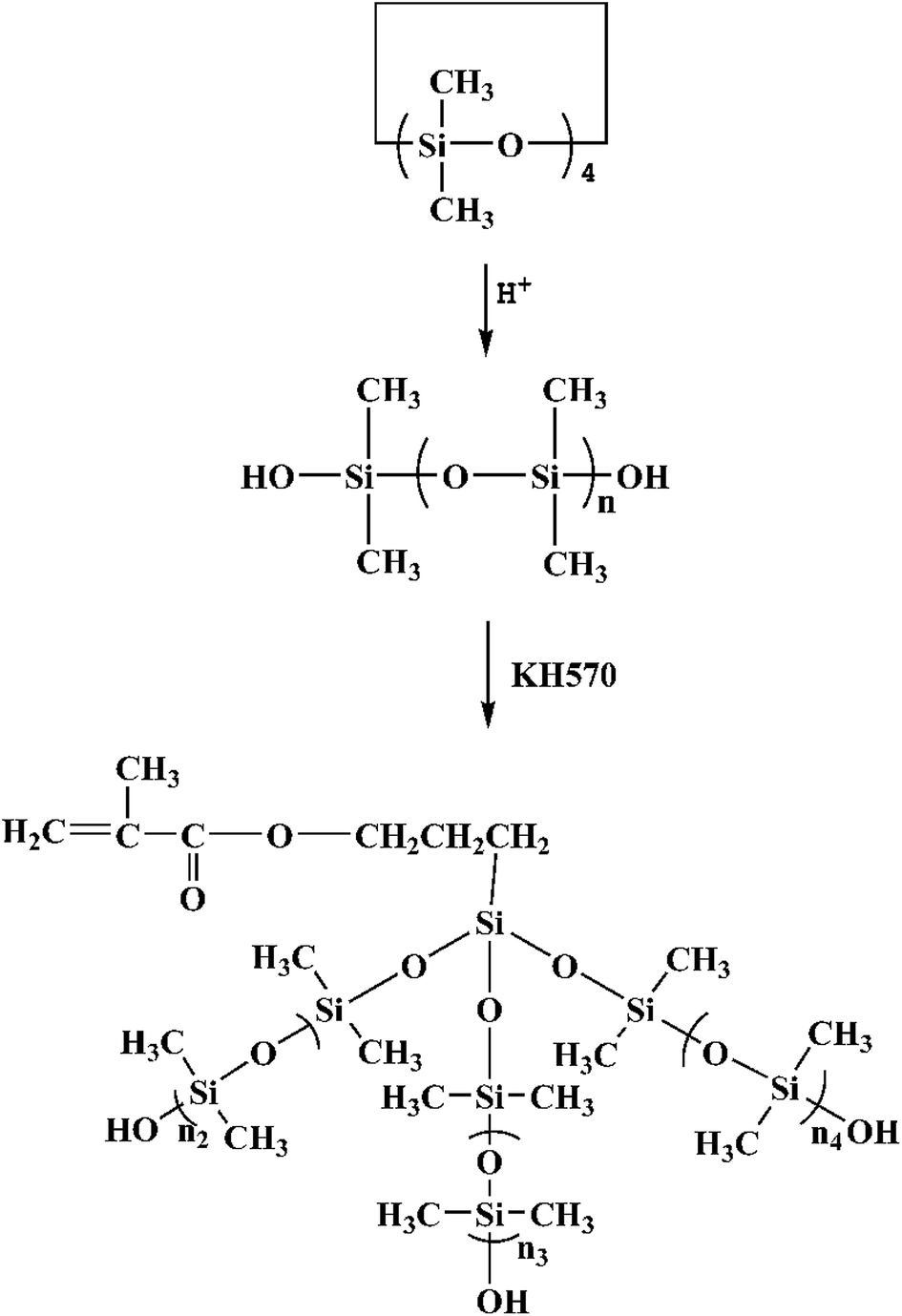
2.3 Synthesis of MATSi
MATSi was synthesized by emulsion polymerization. The distilled water, surfactants (DBSA and OPE) and monomers (D4) were added into a four-necked flask equipped with a thermometer, a reflux condenser, a mechanical stirrer and a nitrogen inlet. DBSA acted as an acid catalyst as well. Nitrogen was added into the flask at first to remove oxygen. The reaction was carried out for 8 h at 80 °C with stirring at about 500 rpm. After being neutralized with NaOH solution to stop the reaction, the final latex of PDMS was obtained and then a certain quantity of KH570 was added. The condensation reaction of PDMS and KH570 was performed at 80 °C for 3 h. The copolymers were precipitated in ethanol and dried in a vacuum drying oven. The product was purified in ethanol and hexane several times to remove the unreacted monomers and surfactants. Through this procedure, the product was obtained and designated as MATSi. The reaction scheme is shown in Scheme 2.
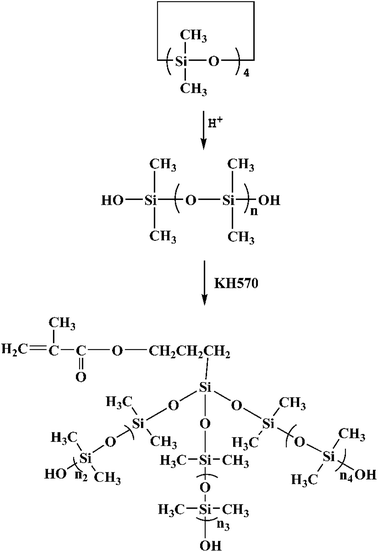 |
| | Scheme 2 Synthesis route of MATSi. | |
2.4 Preparation of the OSPA UV curable films
PA and MATSi were used in relative mass ratios in the range of 100:0 to 90:10 as reported in Table 1. An amount of 5 wt% photoinitiator (Irgacure 1173/triethanolamine = 2:3 w/w) was added to each formulation and stirred for 10 min. Triethanolamine was used to avoid oxygen inhibition during the UV-curing process. Films were then cast onto a glass plate using 5% w/v solutions of the copolymers in methanol by means of a wire-wound applicator. The coated films were laid out at room temperature for at least 5 minutes until the solvent evaporated. The films were then irradiated by a high-pressure mercury lamp (500 W) for 30 s, with a distance of 20 cm from the lamp to the surface of the samples in an air atmosphere. The thickness of the final coating was about 100 μm.
Table 1 Mass ratio of PA and MATSi
| Samples |
PA (%) |
MATSi (%) |
| Pure PA |
100 |
0 |
| OSPA1 |
98 |
2 |
| OSPA2 |
96 |
4 |
| OSPA3 |
94 |
6 |
| OSPA4 |
92 |
8 |
| OSPA5 |
90 |
10 |
2.5 Characterization
The molecular weight and distributions of PA and MATSi oligomer samples were obtained at 25 °C by gel permeation chromatography (GPC) on a Waters 2410 instrument with THF as the solvent (1.0 ml min−1) and polystyrene as the calibration standard.
The FT-IR spectra were obtained with a TENSOR27, Bruker, Germany spectrometer over the range of 400–4000 cm−1. 1HNMR and 29Si NMR were obtained with a 400 MHz Bruker NMR spectrometer using CDCl3 as solvent and tetramethylsilane as the internal reference.
The gel contents of the cured films were determined by measuring the weight loss after a 48 h extraction at 80 °C, according to the standard test method ASTM D2665-84.23 Gel content was calculated as follows:
| | 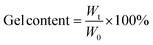 | (1) |
where
W0 is the initial weight of the film, and
Wt is the final weight after extraction.
Flexibility of the UV-cured films was measured according to standard test method (ASTM D522) for elongation of attached coatings, with a conical mandrel apparatus (QTY-32, Shanghai Junda Co., China). The pencil hardness test was conducted on the UV-cured films according to the Standard test method ASTM D2263.
The contact angle measurements were carried out by an optical contact angle meter (Shanghai Zhongchen, China) at room temperature (25 °C), using water and ethylene glycol as pendant drops. Each sample was tested more than 5 times at different locations and averaged readings were used to obtain a reliable value. The surface free energy was calculated by means of a geometric-mean equation, which was described by Owens and Wendt.24 According to Owens and Wendt, the surface energy of a given solid can be determined using an equation applied to two liquids.24,25
| | | (1 + cosθ)γl = 2(γdsγdl)1/2 + 2(γndsγndl)1/2 | (2) |
where
γs and
γl are the surface free energies of the solid and pure liquid, respectively. The superscripts ‘d’ and ‘nd’ represent the dispersive and non-dispersive contributions to the total surface energy, respectively (water:
γl = 72.8 mJ m
−2,
γdl = 21.8 mJ m
−2,
γndl = 51 mJ m
−2; ethylene glycol:
γl = 48 mJ m
−2,
γdl = 29 mJ m
−2,
γndl = 19 mJ m
−2). According to Pinnau and Freeman,
26 the contact angle,
θ, in
eqn (2) was obtained from the following equation:
| | 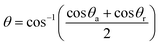 | (3) |
where
θa and
θr are the advancing and receding contact angles, respectively.
The refractive indexes of the coatings were determined by an Abbe refractometer (WAY-2W, Shanghai Electronics Physical Optics Instrument Co., Ltd) at 20 °C.
Scanning electron microscopy (SEM) and energy dispersive spectrometry (EDS) were performed. Cross-section morphologies and elementary distribution of the fracture cured coating films were studied by environmental scanning electron microscopy (Hitachi S-4800 FESEM) with an energy dispersive spectrometer. For SEM inspection, samples were fixed to aluminum stubs with conductive tape prior to coating with ∼20 nm of gold in an Ernest Fullam sputter coater.
The thermal stability of the cured polymeric materials was determined using a thermogravimetric analyzer (TGA, TG209F3, NETZSCH, Germany). The thermogravimetric analysis of selected coatings was carried out at heating rate of 20 °C min−1 under nitrogen atmosphere (flow rate was 30 ml min−1) in the temperature range of 40–600 °C. The differential scanning calorimetry (DSC) thermograms of UV-cured coating samples were obtained using the DSC204 (NETZSCH, Germany) over the range from −60 °C to 120 °C at a heating rate of 10 °C min−1 and held at 120 °C for 5 min to remove the thermal history under N2 atmosphere.
3. Results and discussion
3.1 Synthesis and characterization of OSPA
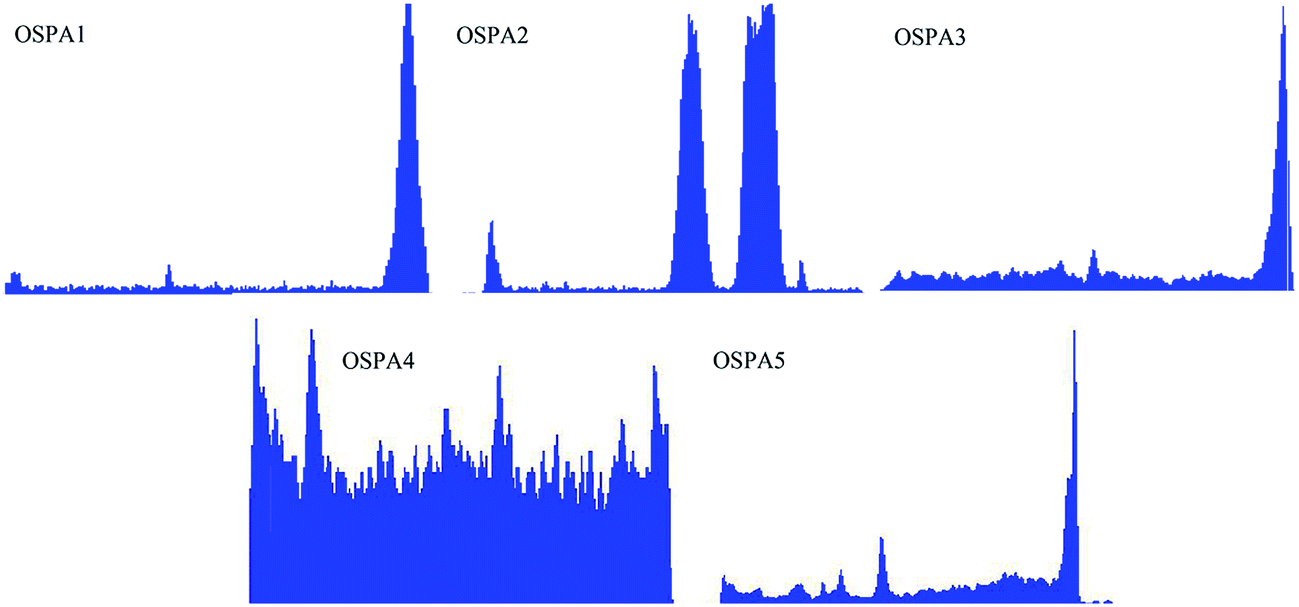
Five UV-curable organosiloxane modified polyacrylates, OSPA1, OSPA2, OSPA3, OSPA4 and OSPA5, were first prepared in this study to study the effects of silicon on the properties of UV-curable composite coatings. The general synthetic scheme for the preparation of the copolymers used in the coating formulations is shown in Scheme 1 for PA and in Scheme 2 for MATSi. GPC, FT-IR, 1HNMR and 29SiNMR spectra were used to determine the structures of the products. The molecular weights and polydispersity indexes of the oligomers were characterized by GPC. The typical molecular weight distributions for PA and MATSi are shown in Fig. 1. As shown in Table 2, the number-average molecular weight of PA and MATSi are 2740 and 1295 g mol−1, respectively. The two mono-modal GPC curves suggest the formation of the two oligomers.
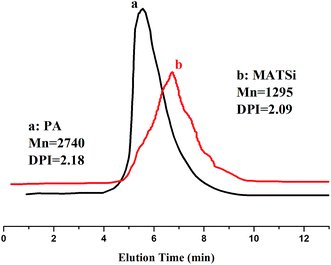 |
| | Fig. 1 GPC traces of PA (a) and MATSi (b). | |
Table 2 Molecular weights of PA and MATSi oligomers
| Samples |
Mna |
PDIb |
|
Mn: the number-average molecular weight, determined by GPC.
PDI: the polydispersity index, determined by GPC.
|
| PA |
2740 |
2.18 |
| MATSi |
1295 |
2.09 |
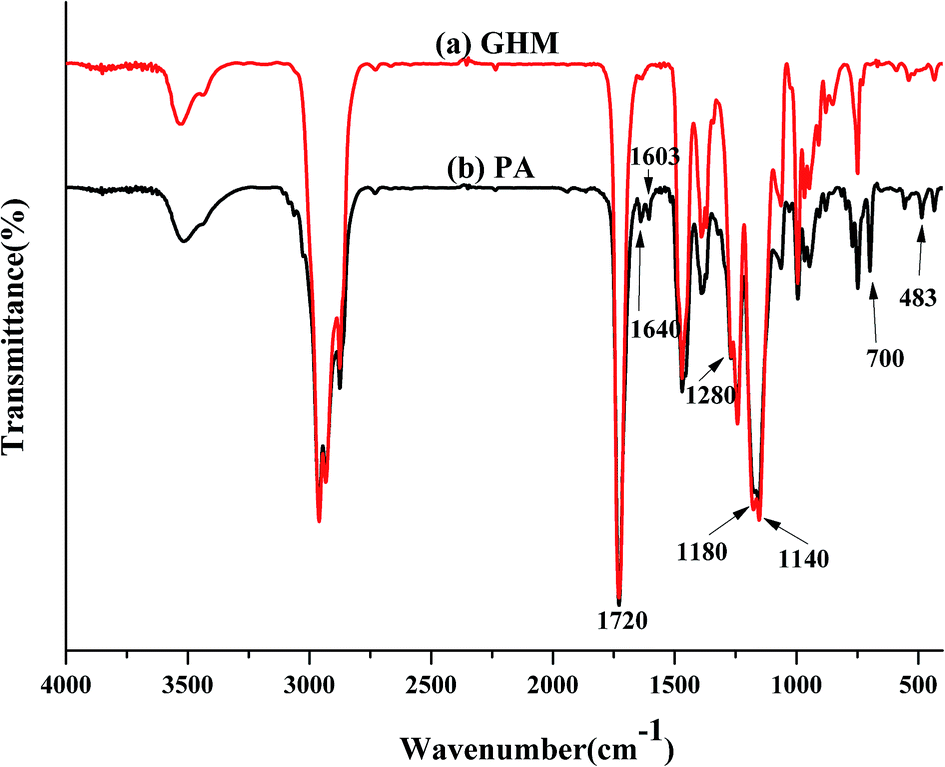
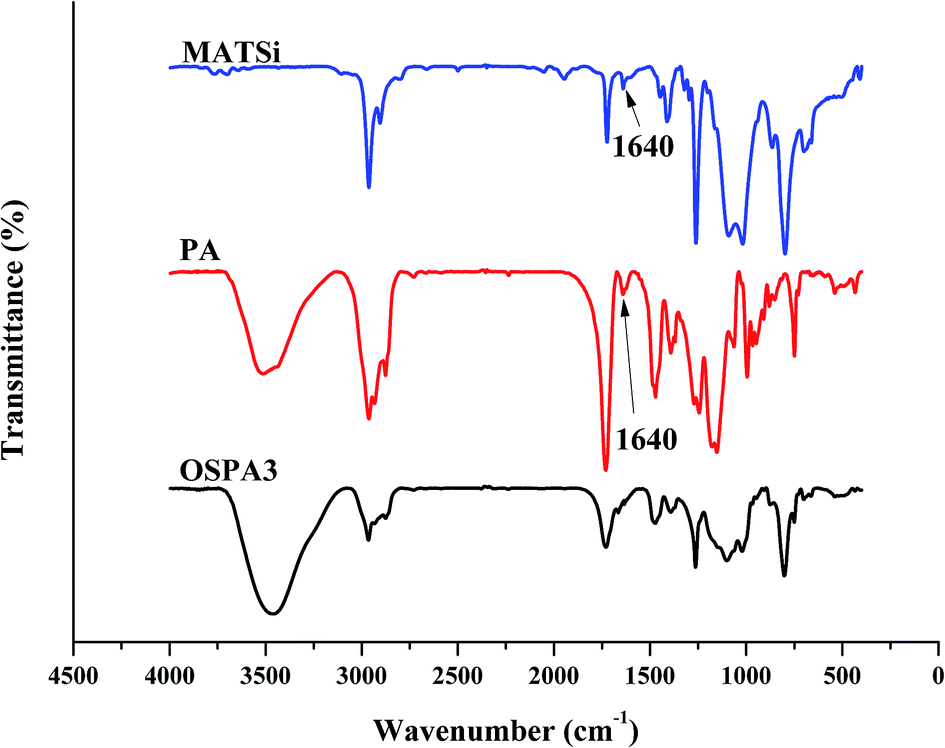
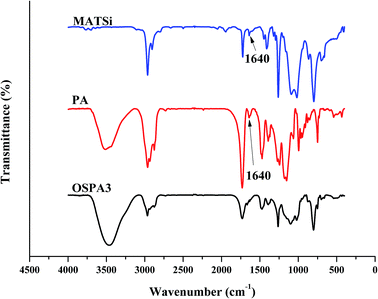
The obtained FT-IR spectra of GHM and PA are reported in Fig. 2. The disappearance of the characteristic absorption peaks of the epoxide group at 910 cm−1 indicates the completion of the reaction. The peaks at 1640 cm−1 (methacrylate double bond) and 700 cm−1 (C–O–H bending) indicate that the epoxy groups have reacted with the addition of MA by a ring-opening addition reaction producing one equivalent of hydroxyl groups. Simultaneously, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C group was introduced into GHM. Absorbance at frequencies characteristic of acrylates (carbonyl CO stretch at 1720 cm−1; C–O stretching bands from 1140 cm−1 to 1180 cm−1 and 1180 cm−1 to 1280 cm−1) are shown.27 Moreover, the characteristic absorption peaks at 1000 and 900 cm−1 for C–O are present in the spectra. The skeletal vibration of the CC group was also present at 483 cm−1.
C group was introduced into GHM. Absorbance at frequencies characteristic of acrylates (carbonyl CO stretch at 1720 cm−1; C–O stretching bands from 1140 cm−1 to 1180 cm−1 and 1180 cm−1 to 1280 cm−1) are shown.27 Moreover, the characteristic absorption peaks at 1000 and 900 cm−1 for C–O are present in the spectra. The skeletal vibration of the CC group was also present at 483 cm−1.
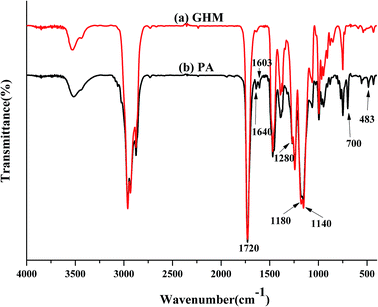 |
| | Fig. 2 FT-IR spectra of GHM and PA. | |
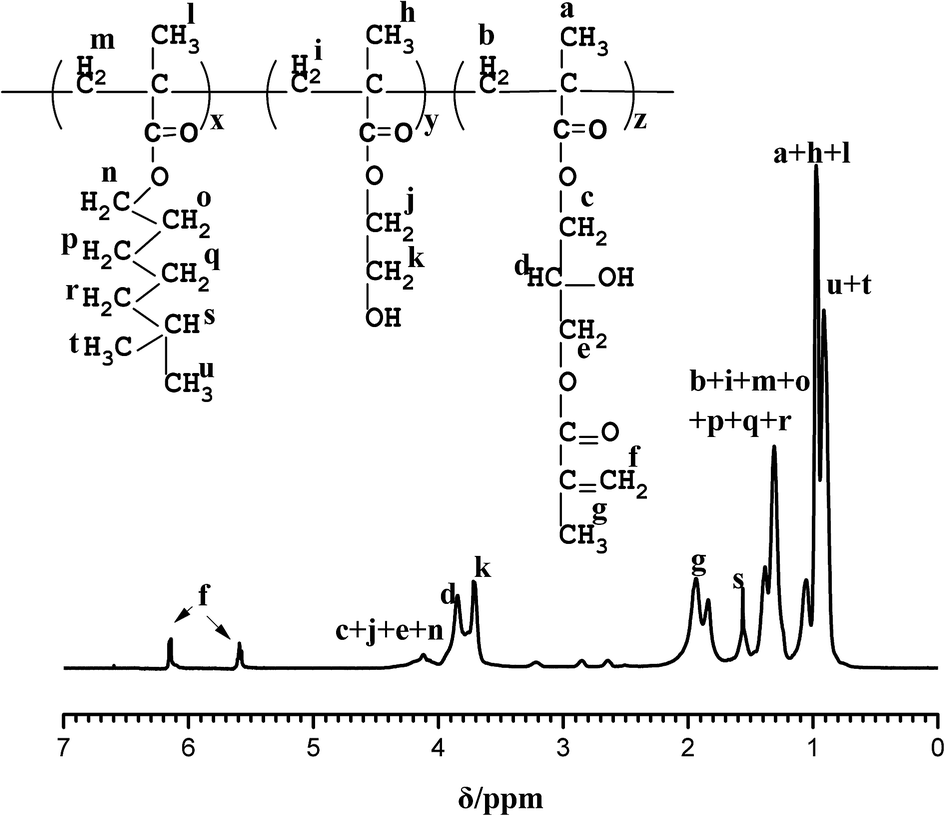
Fig. 3 shows the 1HNMR spectra of PA in CDCl3; peaks in the chemical shift range of 0.7–1.2 ppm and 1.3–1.7 ppm are assigned as –CH3 (a, h, l, u, t) and –CH2 (b, i, m, o, p, q, r), respectively. The proton resonance signals of –CH (s) and –CH3 (g) appear in the regions of 1.5–1.6 ppm and 1.7–2.0 ppm, respectively. The chemical shift of 3.6–3.7 ppm is attributed to protons of HO–CH2 (k) and the chemical shift signals at 3.8–3.9 ppm are attributed to proton of HO–CH (d). The peaks in the chemical shift range of 4.0–4.2 ppm correspond to O–CH2 (c, j, e, n). Peaks at 5.64–5.68 ppm and 6.11–6.18 ppm are assigned as the protons of CCH2 double bonds (f), indicating that the epoxide groups reacted with methacrylic acid and thus CC photosensitive groups were introduced.
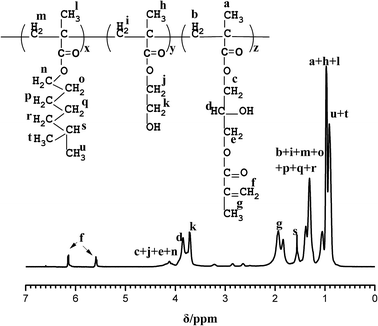 |
| | Fig. 3
1H NMR spectra of PA. | |
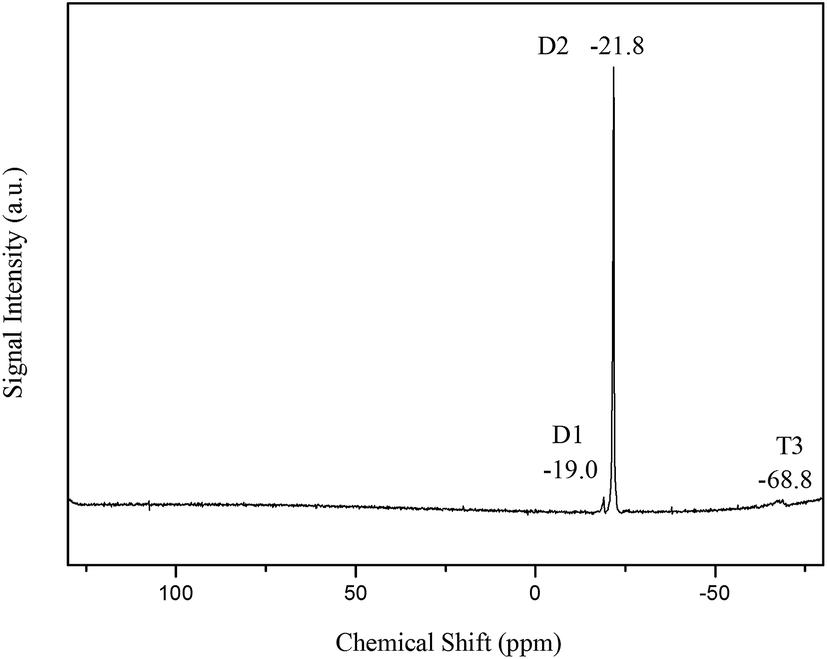
The 29Si NMR signals of mono(T1)-, bi(T2)- and tri(T3)-fold Si–O-linked silicons can be typically observed in the −45 to −50 ppm, −55 to −60 ppm and −65 to −70 ppm regions, respectively.28 In Fig. 4, the 29Si NMR spectra of MATSi are shown. The signal at −68.8 ppm is usually assigned to T3, which represents KH570 after condensation polymerization. The D2 signal appears in the region of −21.8 ppm. It indicates the ring-opening and condensation polymerization reactions of octamethylcyclotetrasiloxane. Poly(dimethylsiloxane) with hydroxyl groups on the end is the product of the reaction. The peak at −19 ppm belongs to the D1 signal of the Si–O group in the MATSi structure. There are no signals in the region of −45 to −50 ppm and −55 to −60 ppm, which indicates that there are no T1 and T2 Si–O-linked silicons in the MATSi structure. It also manifests the complete hydrolysis and condensation reactions of KH570. These results confirm that the MATSi was successfully prepared.
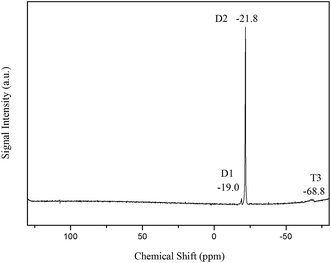 |
| | Fig. 4
29Si NMR spectra of the synthesis of MATSi. | |
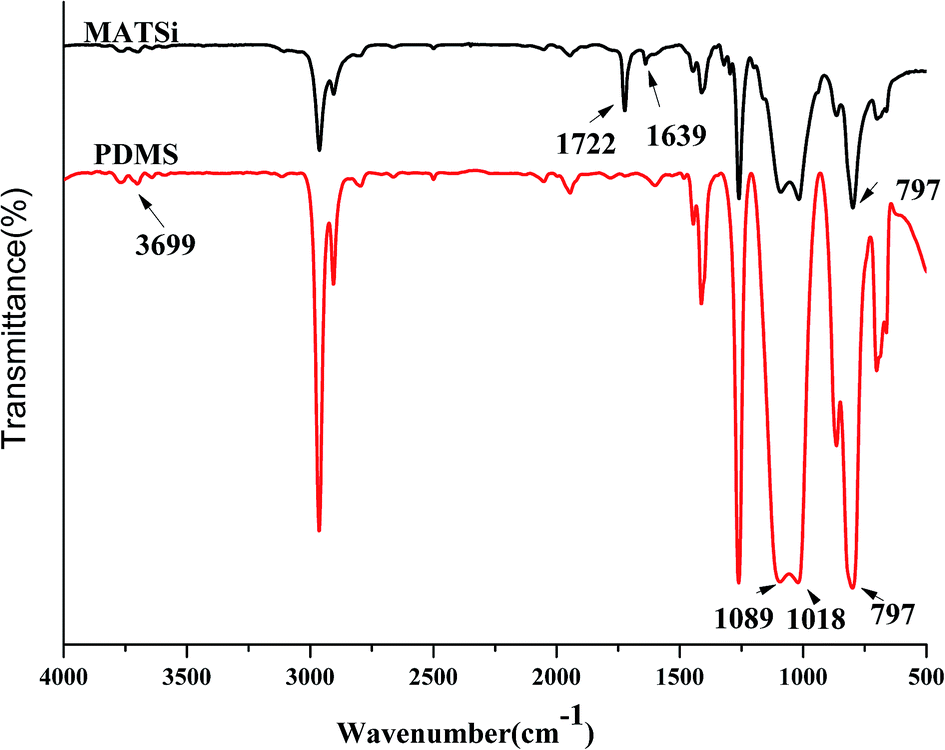
The FT-IR method was also used to confirm the reaction between ring-opened D4 (PDMS) and KH570. From the IR spectra in Fig. 5, it can clearly be observed that PDMS has peaks at 1018 cm−1 and 1089 cm−1, which are the characteristic absorption peaks of Si–O–Si. The Si–CH3 stretching vibration peak at 797 cm−1 was also observed. The peaks of Si–OH at 3699 cm−1 indicate that the ring in D4 was opened and formed polysiloxane. MATSi was produced after PDMS reacted with KH570. The spectra of MATSi shows additional absorption peaks at 1722 cm−1 (CO) and 1639 cm−1 (CC), which represent the reaction between PDMS and KH570.
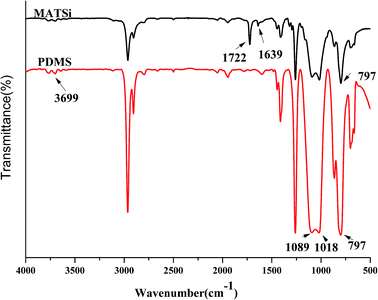 |
| | Fig. 5 FT-IR spectra of the synthesis of MATSi. | |
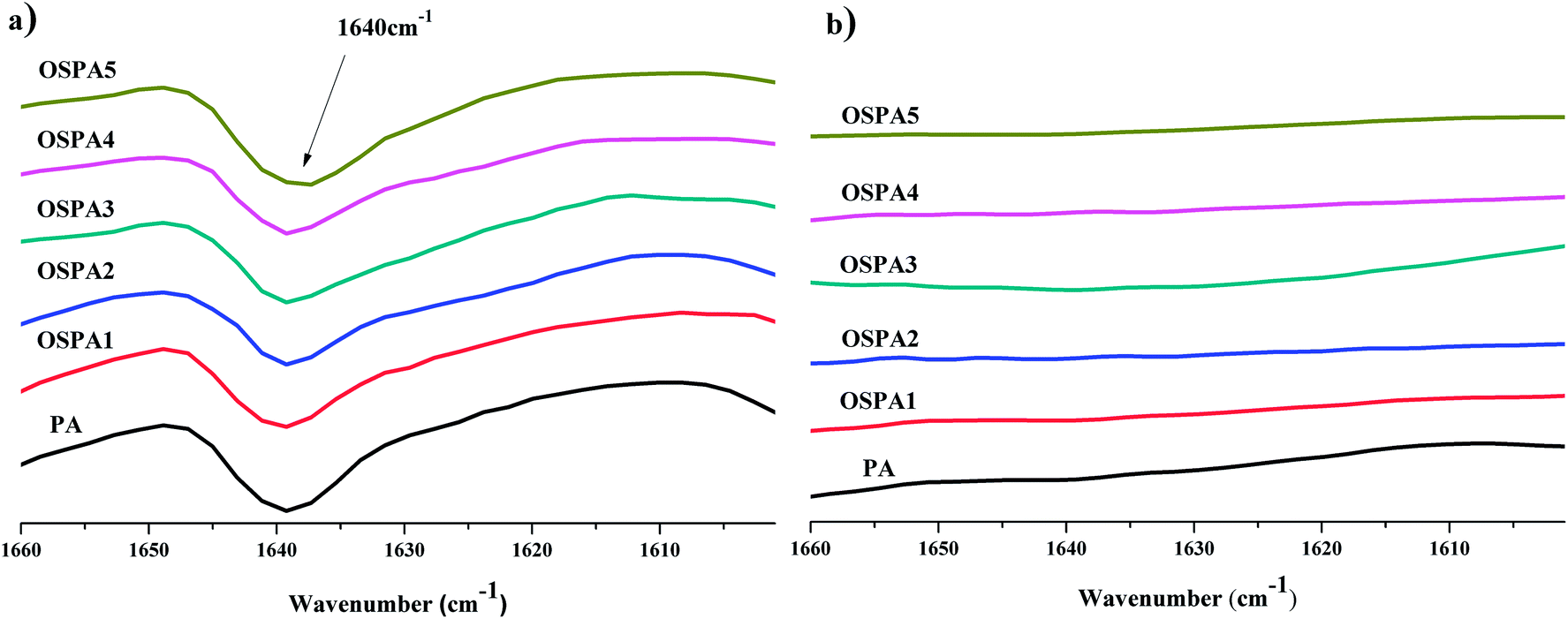
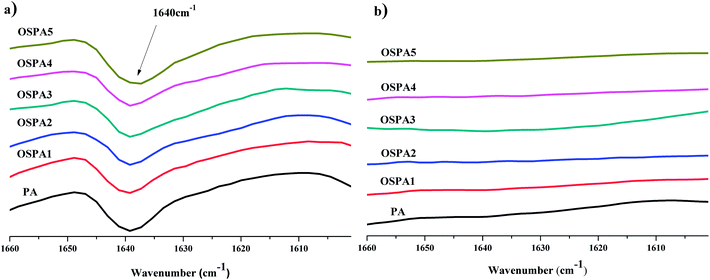
The five UV-cured formulations of PA and MATSi are similar to each other and their IR spectra along with that of OSPA3 are shown in Fig. 6. After irradiation, the characteristic CC vibrations at 1640 cm−1 decreased.
 |
| | Fig. 6 FT-IR spectra of the synthesis of OSPA. | |
The abovementioned characteristic peaks demonstrated that the UV-curable hybrid oligomers based on acrylate containing organosilicon groups were successfully synthesized.
3.2 Degree of conversion in UV curing
FT-IR spectroscopy was used to determine the degree of conversion during the UV crosslinking reaction. The absorption band at 1640 cm−1 due to CC vibration was monitored using FT-IR to determine the degree of conversion by eqn (4).29,30| | 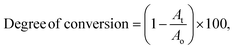 | (4) |
where Ao is the absorption before UV exposure and At is the absorption after UV exposure. The coating formulation was placed on a KBr window and then exposed to UV radiation for 30 s. As shown in Fig. 7, the decrease in peak area of the CC peak at 1640 cm−1 was monitored. Table 3 shows the degree of conversion in coatings after 30 s of exposure to UV radiation; all the OSPA formulations reached high conversion.
 |
| | Fig. 7 FT-IR spectra of the UV-curable film formulations of PA and OSPAs. (a) Before irradiation; (b) after UV-curing. | |
Table 3 Degree of conversion of double bonds in the UV cured coatings
| Coating |
Conversion (%) |
| PA |
95 |
| OSPA1 |
96 |
| OSPA2 |
97 |
| OSPA3 |
99 |
| OSPA4 |
97 |
| OSPA5 |
98 |
3.3 Gel content, flexibility and hardness characterization of the UV-cured coatings
For the purpose of assessing the amount of insoluble parts in the cured films and the mechanical properties of the cured coatings, gel content measurements were conducted. The measured values are summarized in Table 4. The gel content values of all the films are high enough to indicate the nearly complete cross-linked network of the pure PA and composite OSPAs. In terms of flexibility, all the composite OSPA samples passed the 8 mm and 6 mm diameter tests, and most of them passed the 5 mm diameter test, whereas the pure PA sample failed the 8 mm, 6 mm and 5 mm diameter tests. The hardness value of the samples rose from 3H to 6H with the increasing addition of MATSi. The flexibility and hardness tests suggest that the introduction of flexible silicon–oxygen segments enhanced the mechanical properties of the UV-cured coatings.
Table 4 Gel content, flexibility and hardness characterization of the UV-cured coatings
| Samples |
Gel content (%) |
Flexibility |
Pencil hardness |
| 8 mm |
6 mm |
5 mm |
| Pure PA |
97 |
Fail |
Fail |
Fail |
3H |
| OSPA1 |
98 |
Pass |
Pass |
Fail |
5H |
| OSPA2 |
97 |
Pass |
Pass |
Pass |
6H |
| OSPA3 |
98 |
Pass |
Pass |
Pass |
6H |
| OSPA4 |
98 |
Pass |
Pass |
Pass |
6H |
| OSPA5 |
98 |
Pass |
Pass |
Pass |
6H |
3.4 Surface and optics characterization of the cured coatings
To determine the effect of organosilicons on the surface and optical properties of the UV-cured hybrid oligomer films, contact angles and refractive indexes, respectively were investigated. Table 5 presents the contact angles of the five samples. It can be seen that contact angle values of both water and ethylene glycol for the pure PA film are considerably lower than those of the OSPA cured films. With increasing MATSi content, the contact angle values on the OSPA cured film surfaces showed a gradually increasing change. It was found that based on the increase in organosilicon concentration, OSPA tended to be more hydrophobic compared with the virgin PA. Because organosilicon possess low surface energy, they can easily move towards the air–polymer interface causing their enrichment on the coating surface.31,32 The presence of MATSi could therefore lead to a great decrease in the surface free energy, and among these composite films, OSPA5 presented a very low surface free energy value, down to 8.89 mN m−1. Surfaces from mixtures of PA/MATSi with a ratio of 1:0.02 still showed hydrophobicity, although the organosilicon content was low. The results shown in Table 5 indicate that organosilicon contribute to the surface hydrophobicity.
Table 5 Contact angles of the UV-cured coatings with deionized water and ethylene glycola
| |
Surface free energy (mN m−1) |
Contact angle (θ°) |
|
γ
s
|
Deionized water |
Ethylene glycol |
|
γ
s: surface free energy of solid.
|
| PA |
44.28 |
94 |
55.75 |
| OSPA1 |
11.70 |
102.5 |
91 |
| OSPA2 |
12.73 |
108.5 |
91 |
| OSPA3 |
10.36 |
108.5 |
94.5 |
| OSPA4 |
13.27 |
109.5 |
91 |
| OSPA5 |
8.89 |
114 |
99.5 |
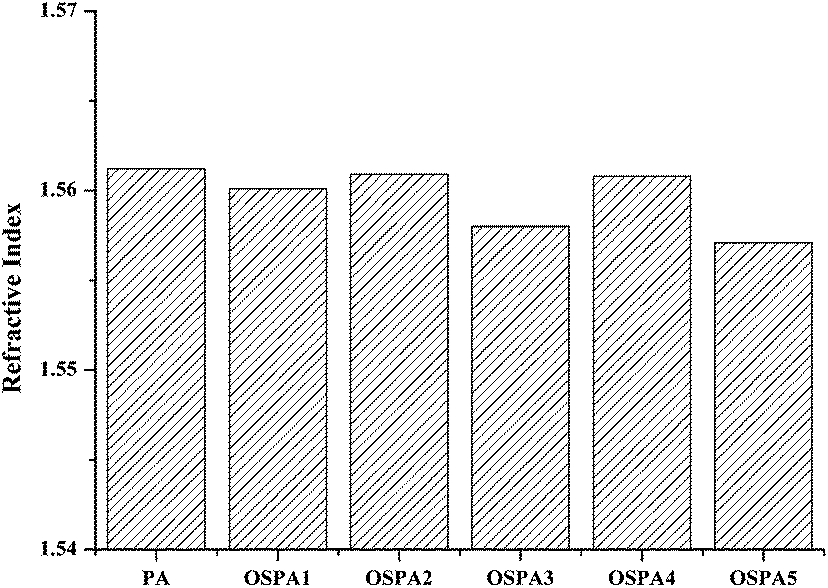
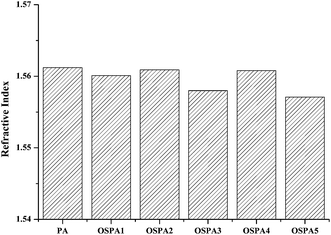
The refractive indexes of the coatings were obtained by an Abbe refractometer at 20 °C, and those obtained for samples OSPA1, OSPA2, OSPA3, OSPA4, and OSPA5 are listed in Fig. 8. As expected, the refractive indexes ranged between 1.5612 and 1.5571 are consistent with expected values for slight amounts of organosilicon in the films. The refractive indexes of the samples showed a very small change with the increase in the organosilicon content, which did not affect the optical properties of the composited material.
 |
| | Fig. 8 Refractive indexes of the UV-cured coatings. | |
3.5 Thermal properties of the cured coatings
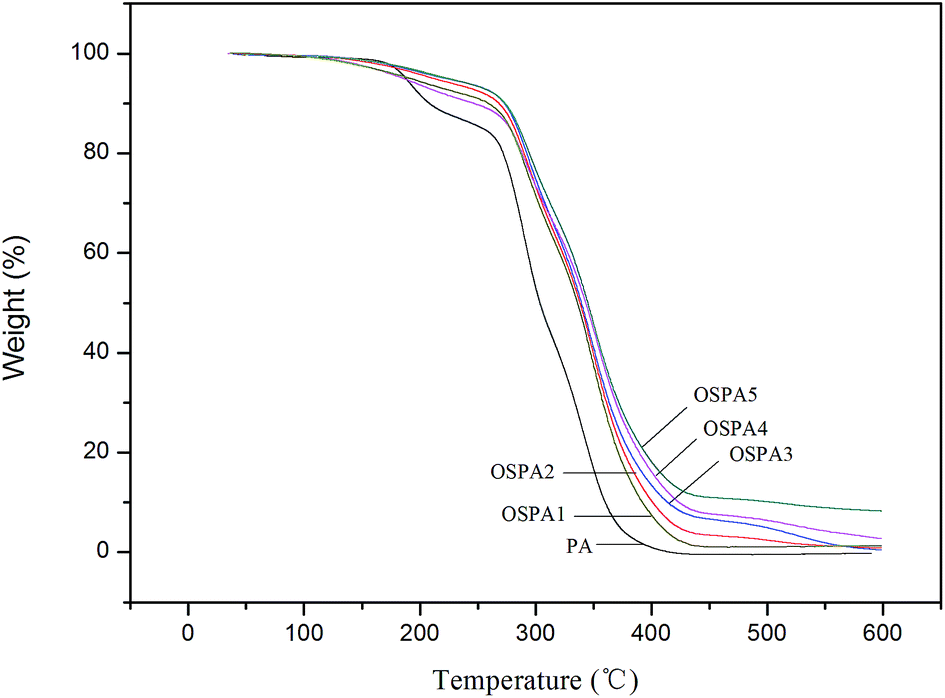
Overall weight loss was observed in composite films with different organosilicon contents. The TGA traces of all cured coatings are included in Fig. 9, and plots of mass loss versus temperature are shown. To gain a better understanding of the degradation behavior of the cured composite coatings, virgin PA film was compared with the composites at three specific degradation temperatures: (a) the temperature of the initial 5% mass loss (T5%); (b) the temperature of the 50% mass loss (T50%) and (c) residual weight percent at 600 °C. The specific degradation data are summarized in Table 6. It shows that the typical onset temperature of the degradation is higher for the composites than for the virgin PA. The thermal stability of the OSPA composites is enhanced relative to that of virgin PA. All the OSPA samples exhibit an apparent higher temperature at 50% weight loss during decomposition, compared with pure PA. The different thermal properties between virgin PA and OSPA may be attributed to some interaction between organosilicon and PA that serves to stabilize the composite. The thermal stability of the composites systematically increases with increasing organosiloxane. The results demonstrate that the incorporated organosiloxane plays an important role during decomposition. The residual silicon-containing compound acts as an insulator and mass transport barrier to the volatile products generated during decomposition. Moreover, compared with virgin PA, the more complex OSPA network reduced the overall rate of evolution of volatiles.
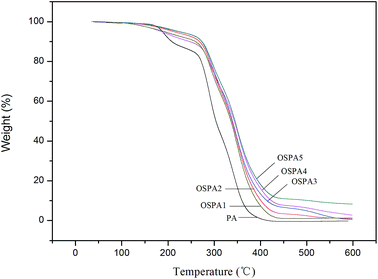 |
| | Fig. 9 TGA curves of the UV-cured coatings. | |
Table 6 Thermal properties of the UV-cured coatings
| Sample |
T
5%
|
T
50%
|
wt% at 600 °C |
T
g
|
| PA |
187.8 |
304.1 |
0 |
15.67 |
| OSPA1 |
220.6 |
339.7 |
0.3 |
22.05 |
| OSPA2 |
211.5 |
338.2 |
0.9 |
24.01 |
| OSPA3 |
190.8 |
335.9 |
1.2 |
27.92 |
| OSPA4 |
183.9 |
343.5 |
2.7 |
27.99 |
| OSPA5 |
224.1 |
345.1 |
8.2 |
27.05 |
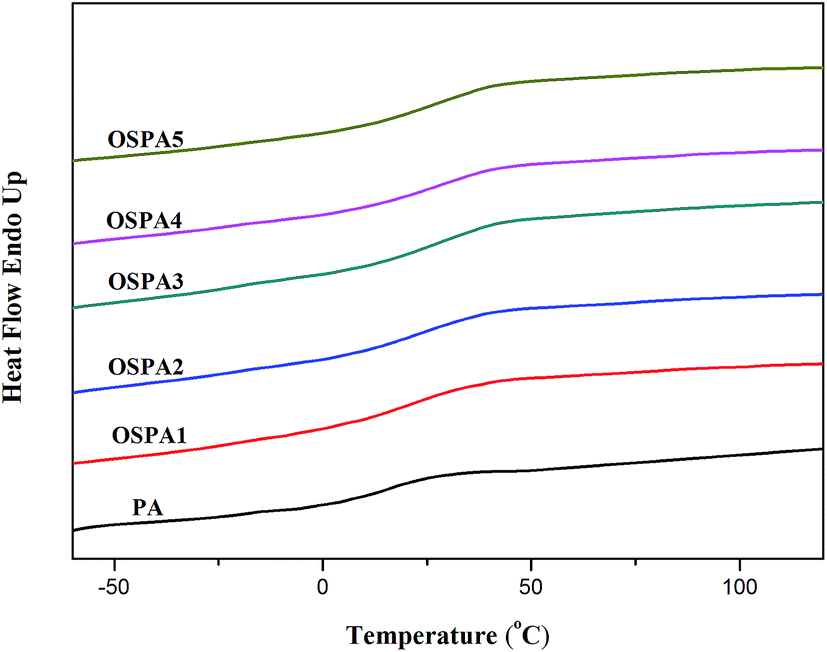
Glass transition temperatures (Tg) of the UV-cured films were investigated by differential scanning calorimetry (DSC). Fig. 10 shows the DSC thermograms of PA and OSPAs. The Tg values of all the samples are summarized in Table 6. There was an increasing trend in Tg values as the amount of MATSi in pure PA increased.
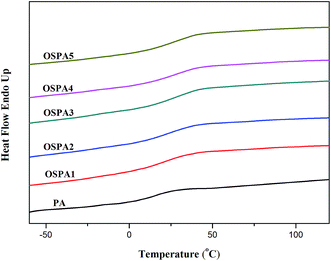 |
| | Fig. 10 DSC thermograms of the UV-cured PA and OSPA coatings. | |
3.6 Micro-morphology of the cured coatings
Fig. 11 shows the fractured-surface microstructure of the cured films cast onto glass slides from a PA/MATSi ratio of 98:2, 96:4, 94:6, 92:8 and 90:10. Sample (a) exhibits a uniform distribution for the network of virgin PA. However, the fractured surfaces of OSPAs showed rougher features than that of virgin PA. These observations also indicate that the distribution of the organosilicon copolymer in virgin PA is not homogeneous. In addition, it can be clearly observed that a certain amount of spheres were enriched closed to the air side surface and there were also spheres in the matrix. This observation is evidence that the silicon-containing groups moved towards the air side surfaces of the cured films. These results can be explained as follows: during the solvent evaporation, silicon-containing groups move towards the top surfaces of the UV-cured formulations owing to their poor compatibility with PA. These observations are in good agreement with the results obtained from the contact angle values. When the content of MATSi increased, the phase separation appeared and became more obvious at the interphase.33–35 It can be inferred that the hydrophobic surfaces may have resulted from the silicon-containing segments in the MATSi groups. The EDS results in Fig. 12 show energy-spectrum scanning from the bottom to the top of the film. It also indicates that the silicon-containing groups assembled on the surfaces of the films, which agree well with SEM and contact angle data. Furthermore, the silicon spheres are always at the ends of the cracks, which means that they could efficiently absorb the energy generated in the fracture process and prevent aggravated fractures.
 |
| | Fig. 11 SEM images of the fractured-surface morphologies of the UV-cured coatings: (a) PA; (b) OSPA1; (c) OSPA2; (d) OSPA3; (e) OSPA4; (f) OSPA5. | |
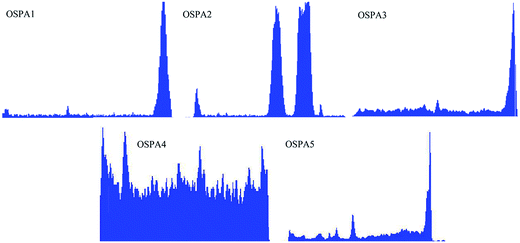 |
| | Fig. 12 EDS images of the fractured surfaces of the UV-cured coatings. | |
4. Conclusion
A novel methacrylate terminated polysiloxane was synthesized by a hydrolysis reaction using octamethylcyclotetrasiloxane and methacryloxy propyl trimethoxysilane. Methacrylate groups were incorporated into the functional polysiloxane oligomer to enhance the compatibility between organosiloxane segments and acrylate film. With the incorporation of organosiloxane, OSPA gained increased thermal and mechanical properties compared to the virgin PA. It was determined that owing to the hydrophobicity of the organosilicon segments, the cured coating films containing PDMS had lower surface free energies and higher thermal degradation temperatures. SEM and EDS studies of the coatings depicted that silicon-containing groups gathered on the air-side surfaces of the cured films. The low cost hydrophobic UV-curable OSPA coatings have a promising combination of physical and mechanical properties, which will lead to potential applications, such as printing inks, paints, adhesives and packaging overcoat films, in the industrial coatings fields.
5. Acknowledements
This study was financially supported by the Program “Tianhe District science and technology plan”.
References
- C. Chen, M. L. Li, Y. J. Gao, J. Nie and F. Sun, RSC Adv., 2015, 5, 33729 RSC.
- D. Knittel and E. Schollmeyer, Polym. Int., 1998, 45(1), 110–117 CrossRef CAS.
- K. J. van den Berg, L. G. J. van der Ven and H. J. W. van den Haak, Prog. Org. Coat., 2008, 61, 110–118 CrossRef CAS PubMed.
- R. Liu, J. Zheng, Z. Li, J. Liu and X. Liu, RSC Adv., 2015, 5(43), 34199–34205 RSC.
-
S. P. Pappas, Radiation curing: Science and Technology, New York, Plenum, 1992 Search PubMed.
- J. H. Lee, R. K. Prud'Homme and I. A. Aksay, J. Mater. Res., 2001, 16(12), 3536–3544 CrossRef CAS.
- H. D. Hwang, C. H. Park, J. I. Moon, H. J. Kim and T. Masubuchi, Prog. Org. Coat., 2011, 72, 663–675 CrossRef CAS PubMed.
-
R. Mehnert, A. Pincus, I. Janorsky, R. Stowe and A. Berejka, UV and EB Curing Technology and Equipment, SITA Technology Ltd., London, 1998, vol. 1 Search PubMed.
- B. Türel Erbay and I. E. Serhatlı, Prog. Org. Coat., 2013, 76, 1–10 CrossRef PubMed.
- O. Chiantore, L. Trossarelli and M. Lazzari, Polymer, 2000, 41(5), 1657–1668 CrossRef CAS.
- P. A. Christensen, A. Dilks, T. A. Egerton and J. Temperley, J. Mater. Sci., 1999, 34(23), 5689–5700 CrossRef CAS.
- S. J. Jeon, J. J. Lee, W. Kim, T. S. Chang and S. M. Koo, Thin Solid Films, 2008, 516, 3904–3909 CrossRef CAS PubMed.
- H. Li, S. Liu, J. Zhao, D. Li and Y. Yuan, Thermochim. Acta, 2013, 573, 32–38 CrossRef CAS PubMed.
- B. U. Ahn, S. K. Lee, S. K. Lee, J. H. Park and B. K. Kim, Prog. Org. Coat., 2008, 62, 258–264 CrossRef CAS PubMed.
- H. D. Hwang and H. J. Kim, React. Funct. Polym., 2011, 71, 655–665 CrossRef CAS PubMed.
- J. P. Lewicki, J. J. Liggat and M. Patel, Polym. Degrad. Stab., 2009, 94, 1548–1557 CrossRef CAS PubMed.
- S. W. Zhang, Z. D. Chen, M. Guo, J. Zhao and X. Y. Liu, RSC Adv., 2014, 4, 30938 RSC.
- M. Alexandre and P. Dubois, Mater. Sci. Eng., R, 2000, 28(1), 1–63 CrossRef.
- M. Lin, F. Chu, A. Guyot, J. L. Putaux and E. Bourgeat-Lami, Polymer, 2005, 46, 1331–1337 CrossRef CAS PubMed.
- H. Li, S. Liu, J. Zhao, D. Li and Y. Yuan, Thermochim. Acta, 2013, 573, 32–38 CrossRef CAS PubMed.
- R. Bai, T. Qiu, F. Han, L. He and X. Li, Appl. Surf. Sci., 2012, 258, 7683–7688 CrossRef CAS PubMed.
- ASTM D2665-84, Standard Specification for Poly(Vinyl Chloride) (PVC) Plastic Drain, Waste, and Vent Pipe and Fittings, reapproved 2009.
- D. Yu, Y. Zhao, H. Li, H. Qi, B. Li and X. Yuan, Prog. Org. Coat., 2013, 76, 1435–1444 CrossRef CAS PubMed.
- D. K. Owens and R. C. Wendt, J. Appl. Polym. Sci., 1969, 12, 1741–1747 CrossRef PubMed.
- T. Ç. Çanak and İ. E. Serhatlı, Prog. Org. Coat., 2013, 76, 388–399 CrossRef PubMed.
-
Formation and modification of polymeric membranes: overview, ed. I. Pinnau and B. D. Freeman, 214th National Meeting of the American-Chemical-Society, Las Vegas, NE, 1997, pp. 1–22 Search PubMed.
- V. V. Krongauz, Thermochim. Acta, 2010, 503–504, 70–84 CrossRef CAS PubMed.
- K. Albert, E. Bayer and B. Pfleiderer, J. Chromatogr., 1990, 506, 343 CrossRef.
- W. Xiao and W. Tu, J. Chem. Eng. Chin. Univ., 2009, 2, 13 Search PubMed.
- I. Rehman, E. H. Andrews and R. Smith, J. Mater. Sci.: Mater. Med., 1996, 7(1), 17–20 CrossRef CAS.
- H. Tavana, F. Simon, K. Grundke, D. Y. Kwok, M. L. Hair and A. W. Neumann, J. Colloid Interface Sci., 2005, 291(2), 497–506 CrossRef CAS PubMed.
- Y. S. Kim, J. S. Lee, Q. Ji and J. E. McGrath, Polymer, 2000, 43(25), 7161–7170 CrossRef.
- Z. Yan, W. Liu, N. Gao, H. Wang and K. Su, Appl. Surf. Sci., 2013, 284, 683–691 CrossRef CAS PubMed.
- M. Sangermano, W. Carbonaro, R. Bongiovanni, R. R. Thomas and C. M. Kausch, Macromol. Mater. Eng., 2010, 295(5), 469–475 CAS.
- W. Liu, S. Ma, Z. Wang, C. Hu and C. Tang, Macromol. Res., 2010, 18(9), 853–861 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.