
Enhanced reduction of graphene oxide by high-pressure hydrothermal treatment
Noel
Díez
,
Agata
Śliwak
,
Stanisław
Gryglewicz
,
Bartosz
Grzyb
and
Grażyna
Gryglewicz
*
Department of Polymer and Carbonaceous Materials, Faculty of Chemistry, Wrocław University of Technology, Gdańska 7/9, 50-344 Wrocław, Poland. E-mail: grazyna.gryglewicz@pwr.edu.pl; Fax: +48 713206506; Tel: +48 713206506
First published on 22nd September 2015
Abstract
A high-pressure assisted hydrothermal treatment is proposed as a facile, green and efficient route for the reduction of aqueous dispersions of graphene oxide. Reactions were performed in an autoclave at mild temperature (180 °C) using only water and nitrogen or hydrogen gas. No further separation or purification of the reduced products was required. X-ray photoelectron spectroscopy, Fourier transform infrared spectroscopy, X-ray diffraction and thermogravimetric analysis revealed that the application of high pressure significantly enhanced oxygen removal. The C/O atomic ratio of the graphene oxide sheets increased from 1.65 to 5.29 upon conventional hydrothermal treatment using autogenous pressure. Higher C/O ratios of 6.35 and 7.93 were obtained for graphene oxides that were reduced under high-pressure of nitrogen and hydrogen, respectively. Specifically, the use of high-pressure hydrogen improved the removal of oxygen double-bonded to carbon. The introduction of covalently bonded heteroatoms, which is commonly observed for the use of reductants such as hydrazine, was not detected. Furthermore, high-pressure reduction led to a better restoration of the sp2 conjugation than was obtained by conventional hydrothermal treatment, as determined by XPS and Raman spectroscopies. These findings illustrate the promise of high-pressure hydrothermal treatments for the eco-friendly mass production of reduced graphene oxide.
Introduction
Graphene is an important focus of materials science due to its unique electronic, thermal, mechanical and optical properties. The increasing demand for graphene in applied research fields such as biomedicine, electronics, composite materials, sensors and energy storage1 has led to extensive study of methods of mass production. Several strategies have been developed for the synthesis of graphene: micromechanical cleavage, chemical vapor deposition, epitaxial growth, liquid phase exfoliation and thermal or chemical reduction of graphene oxide (GO).2–4 Although only the first four methods give rise to graphene with appropriate structure, chemical reduction of GO is the most suitable for large-scale production because of its lower cost and high yield.2 This technique involves three steps. First, (i) graphite is treated with strong oxidants that introduce oxygen functionalities into the carbon nanosheets and increase the interlayer spacing. Because the van der Waals forces between the carbon sheets are decreased after oxidation, the graphite oxide can be (ii) exfoliated by ultrasonication in water or other specific solvents to yield a GO dispersion. The GO is finally (iii) reduced to remove the oxygen and restore the structure and properties of graphene.The widely accepted Lerf–Klinowski model describes GO as carbon planes heavily decorated with epoxy and hydroxyl groups and, to a lesser extent, carbonyl and carboxyl groups at the edges of the nanosheets.5 These oxygen groups can be efficiently eliminated by the use of reducing agents such as hydrazine and its derivatives,6,7 sodium borohydride8 or hydroiodic acid.9 However, these reactants are highly toxic to humans, corrosive or strongly hazardous to the environment.8,10 Moreover, reduction with hydrazine results in the formation of C–N bonds within the reduced carbon sheets; these bonds are difficult to remove and limit the restoration of the sp2 conjugation in the reduced graphene oxide.11 GO has also been successfully reduced using various environmentally friendly methods involving biomolecules and microorganisms,12–16 but these reducing agents contaminate the final product, and further purification is required to obtain reduced nanosheets. Hence, the search continues for simple, cost-effective, and environmentally friendly methods for the mass production of reduced graphene oxide (rGO).
Hydrothermal reduction has been proposed as a green and simple route for the preparation of rGO.17–19 This technique utilizes only superheated water and thus eliminates the incorporation of nitrogen in the rGO and the need for separation/purification steps. Under hydrothermal conditions (temperature < 200 °C and autogenous pressure), superheated water behaves as a strong electrolyte with high diffusion and dielectric constants; the water readily catalyzes bond cleavage of oxygen moieties.17,20 The chemical composition and dispersibility of rGO can be carefully controlled by modification of the operational parameters (initial concentration of GO, temperature, reaction time and pH).18,19 Furthermore, using Raman spectroscopy, Zhou et al.17 have demonstrated that hydrothermal treatment is more effective than hydrazine in repairing the conjugated sp2 network. However, the degree of reduction achieved by hydrothermal treatment (C/O ≈ 5.5) is lower than that obtained using other commonly used reductants.17
In this work, a novel approach for the preparation of hydrothermally rGO with low oxygen content is presented. This method consists of a conventional hydrothermal reduction assisted by the application of nitrogen or hydrogen at high pressure (40 bar). Our results indicate that the removal of oxygen is substantially enhanced by the use of high pressure. In particular, further removal of double-bonded oxygen can be achieved using hydrogen, leading to rGO with lower oxygen content than that obtained by reduction with hydrazine.
Experimental
Synthesis of GO
Coal tar-based graphite supplied by INCAR-CSIC (Oviedo, Spain) was oxidized by Hummers' method. Briefly, 1 g of graphite was added with vigorous stirring to a 250 ml flask containing sulfuric acid (98%, 48 ml) and NaNO3 (1 g). Then, 6 g of KMnO4 was added slowly to the reaction mixture while the flask was held in an ice bath, followed by stirring for 3 h at 35 °C. Then, 200 ml of H2O2 (3%) was slowly added to the flask. Finally, the mixture was decanted, washed with Milli-Q water, and centrifuged several times until a pH-neutral supernatant was obtained. The resulting graphite oxide was treated with 5% HCl and then washed several times with Milli-Q water until achieving a rinse with a pH of 6.5–7. After drying, 200 mg of graphite oxide was dispersed in 200 ml of water and ultrasonicated (40 kHz) for 10 h in a constant temperature bath of 35 °C. The degree of exfoliation was 90%.High-pressure hydrothermal reduction
A 100 ml volume of GO dispersion (0.8 mg ml−1) was introduced into a stainless steel autoclave (4590 Micro Stirred Reactor, Parr Instrument Company). Pressurized nitrogen or hydrogen was injected into the reactor headspace until a pressure of 40 bar was reached. The dispersion was then heated to 180 °C for 0.5, 1, 5 or 24 h with stirring at 200 rpm. Once the temperature reached 180 °C, the inner pressure was ∼80 bar. After the reaction, the autoclave was cooled to room temperature, and the products were dried at 50 °C for 12 h. Samples obtained under high-pressure nitrogen and hydrogen were labeled rGO/N2/t and rGO/H2/t, respectively, where t is the reaction time. GO was also reduced by conventional hydrothermal treatment, during which the inner pressure reached ∼9 bar. Samples obtained in this manner were labeled rGO/t.Characterization of GO and rGOs
The morphologies and height profile of the GO sheets were investigated by atomic force microscopy (AFM) using a Bruker microscope in tapping mode with Nanoscope WSxM software. A drop of GO suspension was diluted to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 and deposited onto mica. After drying, measurements were obtained under ambient conditions. The morphologies of the reduced materials were observed with a Merlin Zeiss field-emission scanning electron microscope (FE-SEM) operating at an accelerating voltage of 3 kV. The crystalline properties of the materials were studied by X-ray diffraction (XRD) using an Ultima IV Rigaku analyzer equipped with a 2 kW X-ray tube (40 kV per 30 mA) using Cu Kα2 radiation (λ = 1.54056 Å). Thermogravimetric analysis (TGA) was performed using a TGA/DSC 1 Analyzer (Mettler Toledo). Five milligrams of sample was placed in a crucible, which was introduced into the thermobalance. The temperature was increased from 20 to 1000 °C at a heating rate of 10 °C min−1 under a nitrogen flow of 100 ml min−1. Raman measurements were obtained using a Bruker Senterra microscope with incident laser light at 532 nm. Measurements were performed at room temperature with a spectral resolution of 2 cm−1 and a laser power of 2.0 mW.
:100 and deposited onto mica. After drying, measurements were obtained under ambient conditions. The morphologies of the reduced materials were observed with a Merlin Zeiss field-emission scanning electron microscope (FE-SEM) operating at an accelerating voltage of 3 kV. The crystalline properties of the materials were studied by X-ray diffraction (XRD) using an Ultima IV Rigaku analyzer equipped with a 2 kW X-ray tube (40 kV per 30 mA) using Cu Kα2 radiation (λ = 1.54056 Å). Thermogravimetric analysis (TGA) was performed using a TGA/DSC 1 Analyzer (Mettler Toledo). Five milligrams of sample was placed in a crucible, which was introduced into the thermobalance. The temperature was increased from 20 to 1000 °C at a heating rate of 10 °C min−1 under a nitrogen flow of 100 ml min−1. Raman measurements were obtained using a Bruker Senterra microscope with incident laser light at 532 nm. Measurements were performed at room temperature with a spectral resolution of 2 cm−1 and a laser power of 2.0 mW.
The elemental compositions of GO and rGOs were determined by X-ray photoelectron spectroscopy (XPS) using a PHI 5000 VersaProbe. The distributions of oxygen groups in GO and rGOs were investigated by deconvolution of the C1s and O1s core-level spectra. The high-resolution spectra of C1s excitation were resolved into six individual peaks ascribed to graphitic carbon (284.5 eV), carbon atoms with sp3 hybridization (285.4 eV), hydroxyl and epoxy groups (286.5 eV), carbonyl or quinone groups (287.6 eV), carboxyl groups (288.9 eV) and a satellite peak corresponding to the π–π* transition in the aromatic systems (290.4 eV). The O1s signal was deconvoluted into three peaks corresponding to quinone (530.7 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups such as ketone and carbonyl (531.8 eV), and hydroxyl and epoxy functionalities (532.8 eV). Curve fittings were performed using an iterative least squares algorithm (CasaXPS software) with a Gaussian–Lorentzian (70/30) peak shape and Shirley background removal. Fourier transform infrared (FTIR) spectra in the region of 750–3750 cm−1 were obtained with a Bruker Vertex 70 spectrometer. Samples were analyzed in attenuated total reflectance mode (ATR-FTIR) using the Pike MIRacle accessory (Pike Technology) equipped with a Ge crystal. Prior to measurements, the samples and detector compartment were evacuated to remove carbon dioxide and water vapor. Final spectra were calculated as the average of 256 measurements with a resolution of 2 cm−1.
O groups such as ketone and carbonyl (531.8 eV), and hydroxyl and epoxy functionalities (532.8 eV). Curve fittings were performed using an iterative least squares algorithm (CasaXPS software) with a Gaussian–Lorentzian (70/30) peak shape and Shirley background removal. Fourier transform infrared (FTIR) spectra in the region of 750–3750 cm−1 were obtained with a Bruker Vertex 70 spectrometer. Samples were analyzed in attenuated total reflectance mode (ATR-FTIR) using the Pike MIRacle accessory (Pike Technology) equipped with a Ge crystal. Prior to measurements, the samples and detector compartment were evacuated to remove carbon dioxide and water vapor. Final spectra were calculated as the average of 256 measurements with a resolution of 2 cm−1.
Results and discussion
The as-exfoliated GO formed a stable brown dispersion in water (Fig. 1a). AFM observations confirmed that the GO dispersion was composed exclusively of monolayers with a thickness of ∼1.2 nm, which is typical of highly oxidized GO. The dispersion was treated under conventional hydrothermal conditions (rGO/t) and with high-pressure nitrogen and hydrogen (rGO/N2/t and rGO/H2/t). After 0.5 h of reaction, rGO/0.5 and rGO/N2/0.5 formed black dispersions (Fig. 1b) due to the partial removal of oxygen groups from the initial GO. The rGO/0.5 and rGO/N2/0.5 dispersions were stable due to the presence of residual negatively charged oxygen moieties in these samples. By contrast, when high-pressure hydrogen or a longer reaction time (1, 5 or 24 h) was applied, a clear solution and a black hydrophobic precipitate were obtained, indicating greater removal of oxygen. Because only water and pressurized gases were used in the hydrothermal processes, pure rGO could be obtained by simple decanting and low-temperature drying. For all hydrothermal methods, FE-SEM analysis of the precipitates after 24 h of reaction revealed an aggregated structure of crumpled nanosheets of rGO (Fig. 2), similar to other hydrothermally rGOs reported in the literature.7,21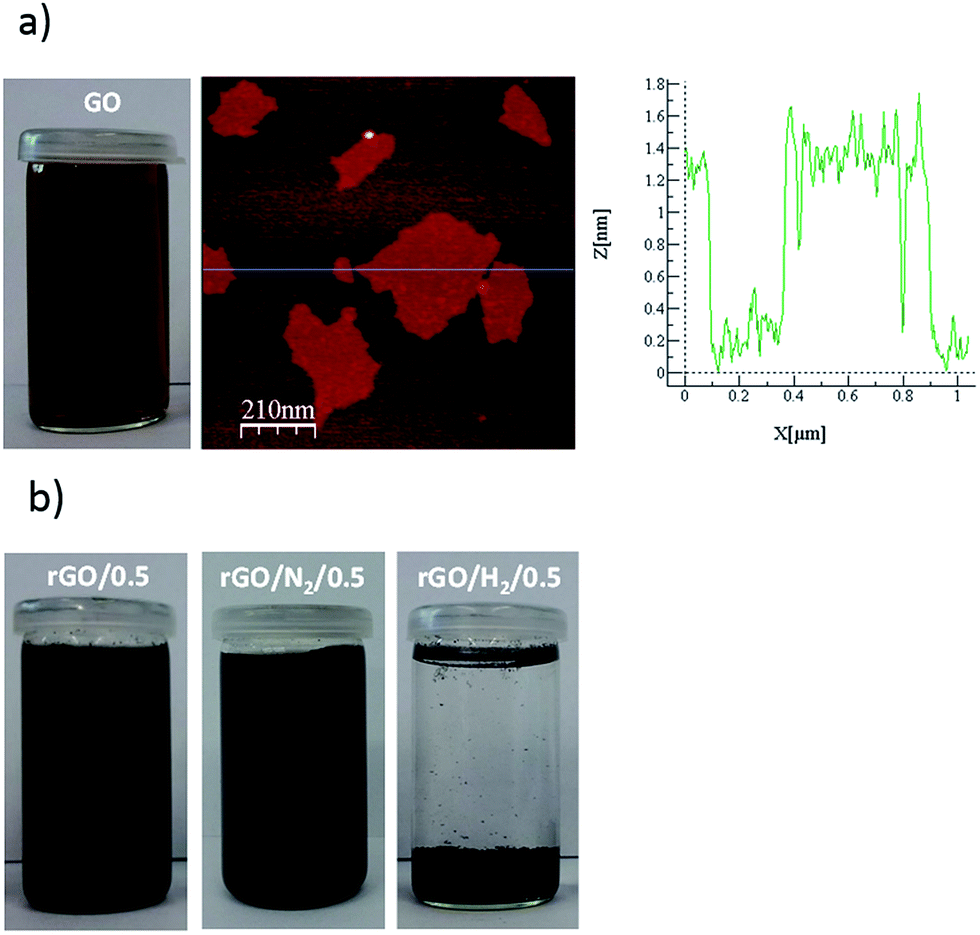 | ||
| Fig. 1 Picture of the GO suspension and AFM image and section analysis of GO (a). Images of as-obtained rGO/0.5, rGO/N2/0.5 and rGO/H2/0.5 (b). | ||
 | ||
| Fig. 2 SEM images of rGO/24, rGO/N2/24 and rGO/H2/24. | ||
The thermal stabilities of GO and the rGO samples were investigated by TGA. Fig. 3a shows the TGA curves of GO and rGOs obtained after 0.5 h of reaction. Two abrupt weight-loss events were observed in the curve of GO. Approximately 12% of the initial weight was lost at temperatures below 120 °C due to desorption of water from the surface. Between 120 and 220 °C, a significant mass loss of 25% of the initial weight occurred. A maximum peak of weight loss was observed at 213 °C, which can be attributed to the decomposition of labile oxygen moieties such as hydroxyl, epoxide and carboxylic groups. At temperatures above 300 °C, slight, steady weight loss due to the removal of more stable oxygen moieties, such as carbonyl groups occurs.22,23 By contrast, samples obtained after 0.5 h of reduction exhibited weight loss of less than 12% below 300 °C, suggesting extensive elimination of oxygen groups upon hydrothermal reduction during the first 30 min of reaction. The plots of rGO/0.5 and rGO/N2/0.5 overlap, while the curve for rGO/H2/0.5 exhibits lower weight loss in the range of 180–450 °C. Profiles from samples reduced after 24 h are depicted in Fig. 3b. Slight differences were observed between the TGA plots of rGO/0.5 and rGO/24, which suggests that the removal of oxygen under autogenous pressure cannot be substantially improved by increasing the reaction time. By contrast, the higher thermal stabilities of rGO/N2/24 and, in particular, rGO/H2/24, are indicative of more extensive removal of oxygen groups upon the application of high pressure.
 | ||
| Fig. 3 TGA and derivative (dotted line) plots for GO and rGOs obtained after 0.5 h (a) and 24 h (b) of reduction. | ||
The crystalline structures of the GO and rGO samples were studied by XRD. The XRD spectrum of GO (Fig. 4a) exhibited a sharp diffraction peak at 2θ = 10.5°, corresponding to an interlayer spacing (d001) of 0.8423 nm, typical of highly oxidized GOs.24 A less intense peak at 42.4°, commonly observed in graphitic substances, was also recorded. The peak at the lower angle disappeared upon hydrothermal treatment, and a new 002 peak occurred at 2θ = 23.4–25.3° (Fig. 4b–d). The broad shape of these 002 peaks suggests that the reduced samples were poorly ordered along the stacking direction and that the rGOs were mainly composed of free-standing graphene sheets. The 002 peaks shifted to higher values of 2θ with increasing reaction time because of the gradual decrease in the interlayer spacing of the rGOs. Greater removal of oxygen under high-pressure reaction conditions could be deduced from the lower d-spacing values calculated for the rGO/N2 and rGO/H2 series. In particular, rGO/H2/24 reached an interlayer spacing of 0.3523 nm, which is significantly lower than the d002 value of 0.37 nm reported for GO reduced with hydrazine.25
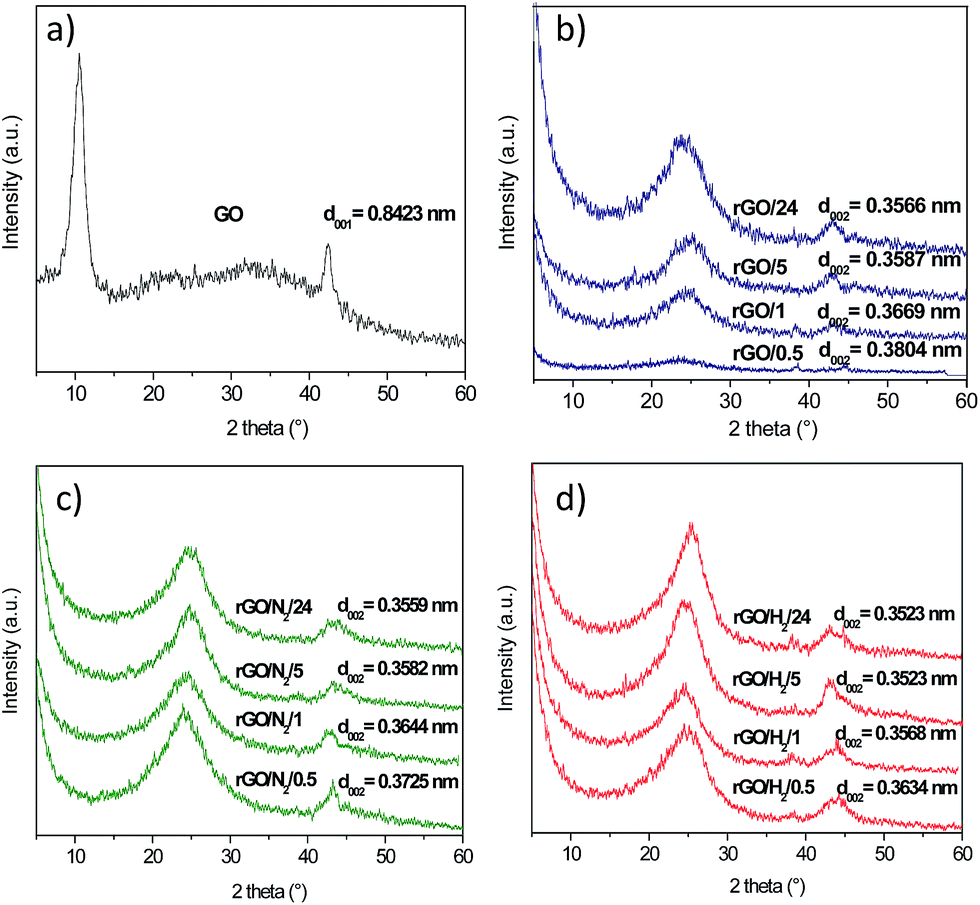 | ||
| Fig. 4 XRD patterns of GO (a), rGO/t (b), rGO/N2/t (c) and rGO/H2/t (d) samples. | ||
The elemental compositions of GO and rGO samples were determined by XPS (Table 1). After 0.5 h of conventional hydrothermal treatment, the oxygen content decreased sharply from 37.8 to 17.7 at%, and after 5 h of reaction, a maximum C/O atomic ratio of 5.29 was achieved (Fig. 5). No further oxygen removal was observed for longer reaction times (i.e., 24 h). However, this limitation was overcome with the introduction of high pressure to the reaction media. The C/O atomic ratio increased progressively with time, reaching values of 6.35 and 7.93 after 24 h of reaction under high-pressure nitrogen and hydrogen, respectively. The oxygen content in rGO/N2/24 and rGO/H2/24 was reduced by an additional 15 and 30%, respectively, compared with rGO/24. rGOs with O/C values of 0.21 and 0.16 were previously obtained by reduction of GO with H2-rich water26 and hydrazine,11,26 respectively. A considerably higher reduction was observed for rGO/H2/24, which achieved an O/C atomic ratio of 0.13. Furthermore, the introduction of heteroatoms (e.g., nitrogen), which is typical of reduction with hydrazine,11 was prevented in the high-pressure assisted reactions, as indicated by the XPS results (Table 1).
| Elemental composition (at%) | C1s peak deconvolution (%) | O1s peak deconvolution (%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C | O | N | Csp2 | Csp3 | C–O | CO |
C(O)OH | π–π* | Quin | CO |
C–O | |
| GO | 62.2 | 37.8 | 0.0 | 22 | 12 | 44 | 17 | 5 | 0 | 3 | 11 | 86 |
| rGO/0.5 | 82.2 | 17.8 | 0.0 | 66 | 10 | 10 | 8 | 4 | 2 | 20 | 32 | 48 |
| rGO/1 | 83.7 | 16.3 | 0.0 | 66 | 11 | 9 | 8 | 3 | 3 | 23 | 34 | 43 |
| rGO/5 | 84.1 | 15.9 | 0.0 | 67 | 12 | 8 | 8 | 3 | 2 | 22 | 36 | 42 |
| rGO/24 | 84.1 | 15.9 | 0.0 | 68 | 11 | 9 | 8 | 3 | 2 | 23 | 36 | 41 |
| rGO/N2/0.5 | 81.5 | 18.5 | 0.0 | 65 | 10 | 10 | 9 | 4 | 2 | 22 | 27 | 51 |
| rGO/N2/1 | 83.7 | 16.3 | 0.0 | 68 | 11 | 9 | 8 | 3 | 1 | 24 | 28 | 48 |
| rGO/N2/5 | 85.8 | 14.2 | 0.0 | 69 | 11 | 8 | 8 | 2 | 2 | 23 | 30 | 47 |
| rGO/N2/24 | 86.4 | 13.6 | 0.0 | 70 | 11 | 7 | 8 | 2 | 2 | 25 | 29 | 46 |
| rGO/H2/0.5 | 83.3 | 16.7 | 0.0 | 67 | 8 | 12 | 9 | 3 | 2 | 24 | 25 | 51 |
| rGO/H2/1 | 85.2 | 14.8 | 0.0 | 68 | 8 | 10 | 9 | 3 | 2 | 27 | 16 | 57 |
| rGO/H2/5 | 87.5 | 12.5 | 0.0 | 71 | 9 | 8 | 8 | 2 | 2 | 24 | 16 | 60 |
| rGO/H2/24 | 88.8 | 11.2 | 0.0 | 73 | 9 | 8 | 6 | 2 | 2 | 29 | 17 | 54 |
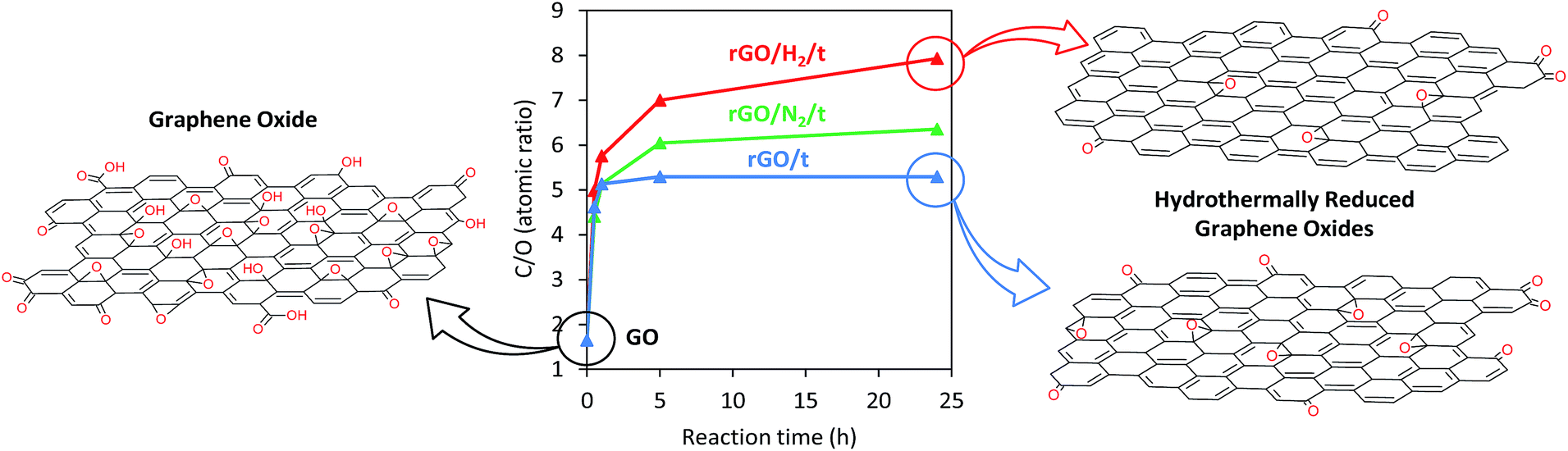 | ||
| Fig. 5 Graph showing the time-correlated C/O atomic ratios of GO and rGO, rGO/N2 and rGO/H2 series as determined by XPS, with proposed structures. | ||
According to Zhou et al.17 superheated water provides large amounts of H+ ions that catalyze the bond cleavage of hydroxyl groups. Under autogenous conditions, the reaction mixture is at boiling point and the bubble nucleation occurs preferentially on the surface of the GO nanosheets. Since the surface of the GO sheets are partially covered with bubbles, the concentration of H+ cations in the surrounding of the GO decreases. The injection of highly pressurized gas inhibits bubbling in the reaction media, which allows a better interaction between the H+ ions in the hydrothermal water and the oxygen groups of GO. The lower oxygen contents observed in the samples treated under high pressure confirm that increasing the reaction pressure is an effective strategy to increase the reduction behaviour of superheated water at mild temperatures. Additionally, H2-rich water is known to have antioxidant properties, being able to reduce some oxygen radicals in biological systems27 and reduce the GO with the same efficiency as hydrazine.26 The effectiveness of H2-rich water as reducing agent is limited by the low equilibrium solubility of hydrogen in water at atmospheric conditions (≈0.0127 cm3 g−1). However, the solubility of hydrogen in water can be increased by applying a high partial pressure of hydrogen in the reactor headspace (it can be incremented by ∼100 times under the experimental conditions used in this work28), and hence higher deoxygenation degrees than those provided by H2-rich water can be expected. In consequence, the very low oxygen contents found in the rGO/H2 samples result from a combined effect of pressurized superheated water and a high concentration of dissolved H2 molecules.
Although rGO with very low or negligible oxygen contents can be achieved by thermal annealing at temperatures as high as 2400 °C,23,29 high pressure hydrothermal treatment can be regarded as a good alternative when the reduction of GO must be conducted in liquid phase or at mild temperature. Hydrothermal treatment conducted in alkaline solution yields stable suspensions of rGO that can be spin-coated into thin films.17 Hydrothermal conditions also allow the reduction of GO assembled on different substrates with low melting point, such as glass or some polymers, which may be valuable for some applications. Furthermore, thermal reduction requires large energy consumption, critical treatment conditions and a continuous flow of an inert gas. In contrast, high pressure hydrothermal reduction provides rGO with low oxygen content operating at mild temperatures and using a reduced amount of nitrogen or hydrogen gas, which is of great interest for the large scale production of rGO.
The high resolution XPS C1s and O1s core level spectra were deconvoluted in order to study the oxygen speciation in the GO and rGO samples (Table 1). Fig. 6a shows the C1s core-level XPS spectra of GO and rGO samples obtained after 24 h of reaction. In accordance with the Lerf–Klinowski model, the spectra of GO primarily consisted of hydroxyl and epoxy groups (286.5 eV) and, to a lesser extent, quinone/carbonyl (287.6 eV) and carboxyl (288.9 eV) functionalities. A substantial decrease in peaks associated with oxygenated carbons was observed for all rGO samples. An intense and well-defined peak at a binding energy of 284.5 eV, attributed to carbon with sp2 hybridization, was observed in all rGOs. The contribution of this peak to the total C1s signal was 68% for rGO/24, 70% for rGO/N2/24, and 73% for rGO/H2/24. These values suggest that the use of high pressure not only promotes additional removal of oxygen but also improves restoration of sp2 conjugation. Three peaks were identified in the O1s core-level spectra (Fig. 6b) that corresponded to quinone (530.7 eV), oxygen double-bonded to carbon (531.8 eV), and oxygen single-bonded to carbon (532.8 eV).30 A sharp decrease in the intensity of the peak at 532.8 eV revealed that oxygen single-bonded to carbon is preferentially removed upon hydrothermal treatment, which is in agreement with the reaction mechanism proposed by Zhou et al.17 The distribution of residual oxygen groups in the rGO/24 and rGO/N2/24 samples is comparable, while the relative intensity of the CO peak is lower in the case of rGO/H2/24. This is indicative of a new reduction mechanism provided by the presence of dissolved hydrogen, which is complementary to that provided by superheated water. Presumably, hydrogen molecules could react with CO bonds to yield hydroxyl groups, which were then effectively removed under hydrothermal conditions. Further investigation should be carried out to clarify the role of dissolved hydrogen in the hydrothermal reduction of GO.
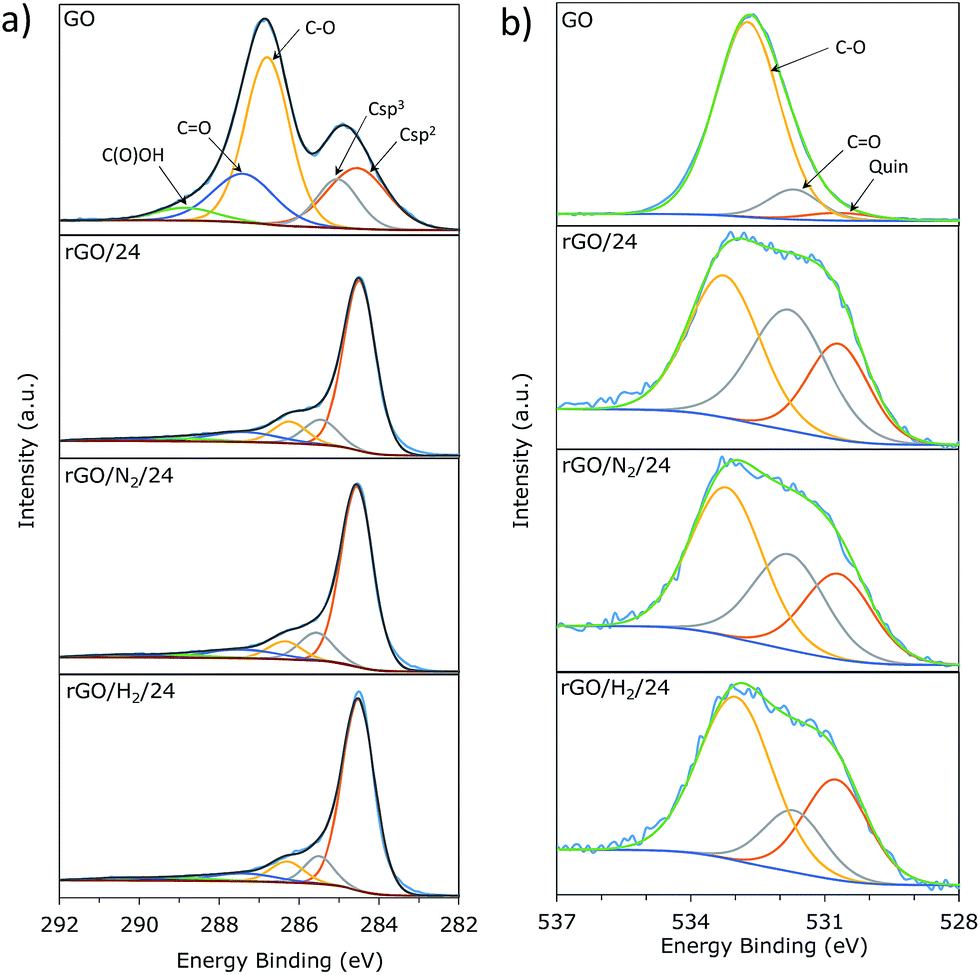 | ||
| Fig. 6 Deconvolution of C1s (a) and O1s (b) core-level XPS spectra of GO, rGO/24, rGO/N2/24 and rGO/H2/24. | ||
The ATR-FTIR spectra of GO and reduced samples after 24 h are presented in Fig. 7. In all reduced samples the band associated with the O–H stretching vibration in hydroxyl groups (3000–3500 cm−1) and the bands ascribed to C–O bending and stretching vibrations in hydroxyl groups (1300–1400 cm−1 and 1055 cm−1, respectively22) disappear, suggesting an efficient removal of these oxygenated groups by hydrothermal treatment. The intensity of the narrow band at 1730 cm−1, corresponding to the CO stretching vibrations from carboxyl groups, decreased after conventional hydrothermal treatment and almost disappeared after high-pressure treatment, particularly when high-pressure hydrogen was used. This observation is in agreement with the XPS results and confirms the preferential removal of double-bonded oxygen under high-pressure assisted reductions. The broad band observed at 1220 cm−1 for all reduced samples was assigned to residual epoxy and quinone groups and decreased in intensity in the high-pressure treatments. The narrow band observed at 1620 cm−1 in GO is attributable to the stretching vibration of CC in unoxidized graphitic domains and the bending vibration in molecules of intercalated water, although a small contribution from ketone groups cannot be discarded.22 This band was much more intense in the reduced samples, indicating the restoration of the conjugated carbon structure.
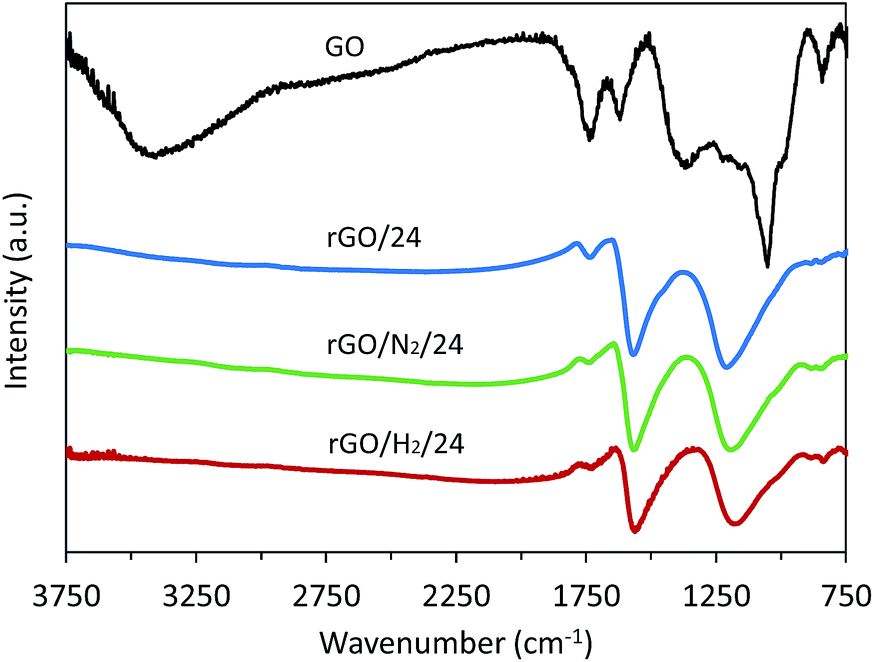 | ||
| Fig. 7 FTIR spectra of GO, rGO/24, rGO/N2/24 and rGO/H2/24. | ||
The carbon structure of the rGOs was further examined by Raman spectroscopy. The Raman spectra of samples obtained after 24 h of reduction are presented in Fig. 8. The band at 1593 cm−1 (G band) originates from the first-ordering scattering of E2g phonons of the graphitic domains. The band at 1352 cm−1 (D band) arises from a breathing mode of k-point phonons of Ag1 symmetry and is attributable to the structural defects in the graphene nanosheets. Thus, the AD/AG intensity ratio can be used to interpret the disorder/order of the graphene nanosheets. A lower AD/AG ratio was observed for the samples reduced under high pressure (AD/AG = 1.72 and 1.79 for rGO/N2/24 and rGO/H2/24, respectively) than for the sample reduced using conventional hydrothermal treatment (AD/AG = 1.86). The lower AD/AG ratios observed for rGO/N2/24 and rGO/H2/24 indicate greater recovery of the graphitic structure due to greater removal of oxygen, which is in agreement with the results obtained from XPS deconvolution.
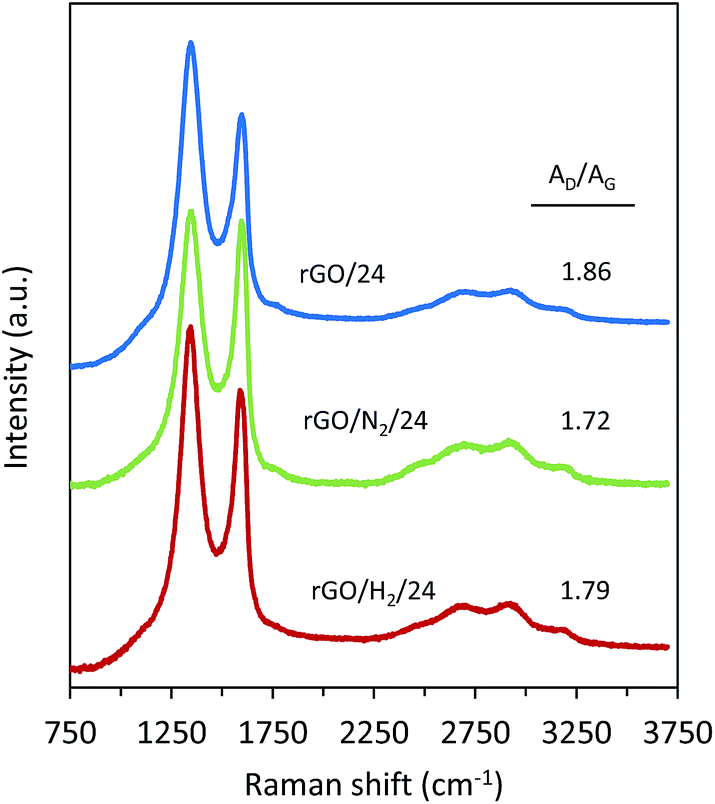 | ||
| Fig. 8 Raman spectra of rGO/24, rGO/N2/24 and rGO/H2/24. | ||
Conclusions
A high-pressure hydrothermal reduction was applied to an aqueous dispersion of GO and compared with a conventional hydrothermal treatment conducted under autogenous pressure. The use of high-pressure (40 bar) nitrogen or hydrogen significantly enhanced the deoxygenation efficiency of hydrothermal treatments. Specifically, the removal of oxygen functionalities was increased by 15 and 30% with the use of nitrogen and hydrogen, respectively. In particular, a C/O atomic ratio of ≈8 was obtained for rGO/H2/24, which is higher than those obtained by treatment with hazardous and commonly used reductants. Moreover, this technique does not introduce nitrogen moieties into the graphene sheets, favouring improved restoration of the sp2 conjugated structure. These findings indicate the potential of high-pressure hydrothermal treatment as a simple and green technique for the mass production of rGO.Acknowledgements
The research leading to these results has received funding from the European Union's Research Fund for Coal and Steel (RFCS) research program under grant agreement RFCR-CT-2013-00006. The authors would thank Integrated Laboratory of Advanced Materials Research and Engineering of Faculty of Chemistry in Wrocław University of Technology for ATR-FTIR and Raman measurements.References
- A. K. Geim, Science, 2009, 324, 1530 CrossRef CAS PubMed.
- S. Pei and H. M. Cheng, Carbon, 2012, 50, 3210 CrossRef CAS.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun and S. de, et al. , Nat. Nanotechnol., 2008, 3, 563 CrossRef CAS PubMed.
- Z. Wang, J. Liu, W. Wang, H. Chen, Z. Liu and Q. Yu, et al. , Chem. Commun., 2013, 49, 10835 RSC.
- H. Y. He, J. Klinowski, M. Forster and A. Lerf, Chem. Phys. Lett., 1998, 287, 53 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and S. Rodney, Carbon, 2007, 45, 1558 CrossRef CAS.
- H. J. Shin, K. K. Kim, A. Benayad, S. M. Yoon, H. K. Park, I. S. Jung, M. H. Jin, H.-K. Jeong, J. M. Kim, J.-Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987 CrossRef CAS.
- S. Pei, J. Zhao, J. Du, W. Ren and H. M. Cheng, Carbon, 2010, 48, 4466 CrossRef CAS.
- J. Zhao, S. Pei, W. Ren, L. Gao and H. Cheng, ACS Nano, 2010, 4, 5245 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228 RSC.
- M. J. Fernández-Merino, L. Guardia, J. I. Paredes, S. Villar-Rodil, P. Solís-Fernández, A. Martínez-Alonso and J. M. D. Tascón, J. Phys. Chem. C, 2010, 114, 6426 Search PubMed.
- C. Zhu, S. Guo, Y. Fang and S. Dong, ACS Nano, 2010, 4, 2429 CrossRef CAS PubMed.
- O. Akhavan, M. Kalaee, Z. S. Alavi, S. M. A. Ghiasi and A. Esfandiar, Carbon, 2012, 50, 3015 CrossRef CAS.
- M. Agharkar, S. Kochrekar, S. Hidouri and M. A. Azeez, Mater. Res. Bull., 2014, 59, 323 CrossRef CAS.
- I. Roy, D. Rana, G. Sarkar, A. Bhattacharyya, N. R. Saha, S. Mondal, S. Pattanayak, S. Chattopadhyay and D. Chattopadhyay, RSC Adv., 2015, 5, 25357 RSC.
- Y. Zhou, Q. Bao, L. A. L. Tang, Y. Zhong and K. P. Loh, Chem. Mater., 2009, 21, 2950 CrossRef CAS.
- N. J. Ding, Y. B. Liu, N. Y. Yuan, G. Q. Ding, Y. Fan and C. T. Yu, Diamond Relat. Mater., 2012, 21, 11 CrossRef.
- C. Bosch-Navarro, E. Coronado, C. Marti-Gastaldo, J. Sanchez-Royo and M. G. Gomez, Nanoscale, 2012, 4, 3977 RSC.
- H. Chen, Z. Song, X. Zhao, X. Li and H. Lin, RSC Adv., 2013, 3, 2971 RSC.
- C. Nethravathi and M. Rajamathi, Carbon, 2008, 46, 1994 CrossRef CAS.
- M. Acik, G. Lee, C. Mattevi, A. Pirkle, R. M. Wallace, M. Chhowalla, K. Cho and Y. Chabal, J. Phys. Chem. C, 2011, 115, 19761 CAS.
- C. Botas, P. Álvarez, C. Blanco, R. Santamaría, M. Granda, M. D. Gutiérrez, F. Rodríguez-Reinoso and R. Menéndez, Carbon, 2013, 52, 476 CrossRef CAS.
- B. Yuan, Ch. Bao, X. Qian, P. Wen, W. Xing and L. Song, Mater. Res. Bull., 2014, 55, 48 CrossRef CAS.
- S. Park, J. H. An, I. W. Jung, R. D. Piner, S. J. An, X. Li, A. Velamakanni and R. S. Ruoff, Nano Lett., 2009, 9, 1593 CrossRef CAS PubMed.
- O. Akhavan, R. Azimirad, H. T. Gholizadeh and F. Ghorbani, Int. J. Hydrogen Energy, 2015, 40, 5553 CrossRef CAS.
- I. Ohsawa, M. Ishikawa, K. Takahashi, M. Watanabe, K. Nishimaki and K. Yamagata, et al. , Nat. Med., 2007, 13, 688 CrossRef CAS PubMed.
- V. I. Baranenko and V. S. Kirov, At. Energy, 1989, 66, 30 CrossRef.
- S. Pei and H. M. Cheng, Carbon, 2012, 50, 3210 CrossRef CAS.
- X. Fan, Ch. Yu, J. Yang, Z. Ling and J. Qiu, Carbon, 2014, 70, 130 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |