
New 13-pyridinealkyl berberine analogues intercalate to DNA and induce apoptosis in HepG2 and MCF-7 cells through ROS mediated p53 dependent pathway: biophysical, biochemical and molecular modeling studies†
Sabyasachi Chatterjeea,
Sumana Mallickb,
Franco Buzzettic,
Gaetano Fiorilloc,
Tanjia Monir Syedac,
Paolo Lombardic,
Krishna Das Sahab and
Gopinatha Suresh Kumar*a
aBiophysical Chemistry Laboratory, Organic and Medicinal Chemistry Division, CSIR-Indian Institute of Chemical Biology, 4, Raja S. C. Mullick Road, Kolkata 700 032, India. E-mail: gskumar@iicb.res.in; Fax: +91 33 2472 3967; Tel: +91 33 2472 4049
bCancer Biology and Inflammatory Disorder Division, CSIR-Indian Institute of Chemical Biology, Kolkata 700 032, India
cNaxospharma srl, Via G. Di Vittorio, 70, 20026 Novate Milanese, MI, Italy
First published on 15th October 2015
Abstract
A new series of 13-pyridinealkyl berberine analogues was synthesized and their DNA binding efficacy studied by employing spectroscopic, calorimetric and molecular modeling techniques. Analogues with more than one CH2 group showed better intercalative binding than berberine. The analogue with one CH2 group bound DNA weaker than berberine. The binding of the analogue with single CH2 group was entropy driven, while those with more than one CH2 group was favoured by both entropy and enthalpy changes. Higher salt concentration and temperature destabilized the binding. A larger contribution from non-polyelectrolytic forces to the Gibbs energy and the involvement of strong hydrophobic interactions were inferred. Molecular modeling pin pointed the specific binding site and the non-covalent interactions in the association. The best DNA binding analogue (BER5) inhibited the growth of hepatocellular and breast carcinoma most efficiently. It induced apoptosis in HepG2 and MCF-7 cells with externalization of phosphatidylserine and reactive oxygen species generation with accumulation of cells in the G0/G1 phase. Furthermore, up regulation of p53 and p21 indicated the role of p53 in BER5 mediated apoptosis. The results suggested that 13-pyridinealkyl berberine analogues intercalated to DNA much stronger than berberine, the chain length of the linker plays an important role for the binding, and they induced ROS mediated apoptosis in HepG2 and MCF-7 cells by p53 modulation.
Introduction
In cellular systems, DNA is frequently targeted by low molecular weight ligands. DNA targeted therapeutic agents can interfere with numerous biological functions, the most important being the transcription and replication of DNA. Such interference can effectively retard or prevent cell division leading to chemotherapeutic action. A large spectrum of such DNA targeting drugs have been studied since the discovery of the structure of DNA. Some like doxorubicin, dactinomycin, quarfloxin, mitoxantrone, mitomycin C, and cisplatin have been clinically employed, while many others are under different stages of investigation and clinical trials.Berberine (Scheme 1a), a natural isoquinoline plant alkaloid of the structural class protoberberines, has many remarkable biological properties, the most potent albeit less investigated being its anticancer activity. A number of interesting studies, both in vitro and in vivo, on the antineoplastic activity of berberine have been reported in the literature that provide potential leads to promising futuristic clinical application for this alkaloid.1–6 Nucleic acids are believed to be the cellular targets of berberine in manifesting its anticancer activity and consequently many structural studies in this direction have been performed. An oligonucleotide–berberine complex has been recently crystallized and the structure solved.7 The DNA binding studies have also been reviewed extensively in many recent articles.8,9 Berberine has the unique distinction of exhibiting remarkable adenine–thymine base pair specificity unlike most other anticancer drugs that are specific to guanine–cytosine sequences.10 But the critical limitations of berberine to be employed as a pharmaceutical preparation are inter alia its poor solubility, low bioavailability and moderate nucleic acid binding affinity. To circumvent these problems attempts have been made to form inclusion complexes, nanoformulations, etc., that have met with only limited success.11–13 It has thus become apparent that more effective derivatives with enhanced DNA and/or RNA binding efficacy are essential for harnessing the anticancer activity of berberine. Recently a number of berberine (BER) analogues with aromatic moieties incorporated at the 13-position were reported to be more effective than berberine in exerting anticancer activity delaying the development of tumours.5,14,15
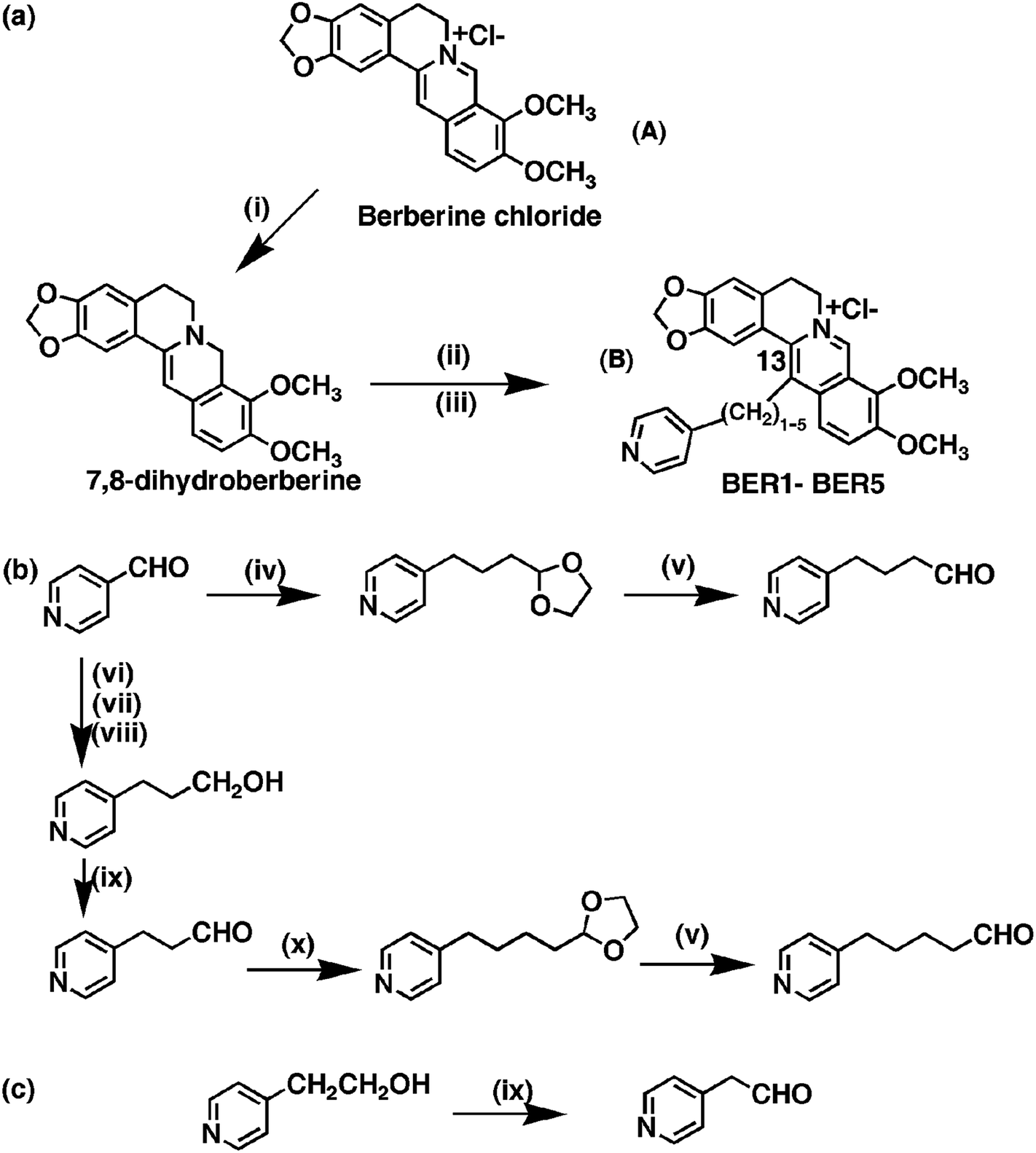 | ||
Scheme 1 (i) NaBH4, pyridine, rt, 1 h, (ii) Py(CH2)0–4CHO, 80% aq. EtOH, AcOH, 85 °C, (iii) 2N HCl, rt, 2 h. (iv) 2-(1,3-Dioxolan-2-yl)ethyltriphenylphosphonium bromide, t-BuOK, THF, rt, (v) 60% aq TFA, rt, (vi) Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCOOEt, 150 °C, 2 h, (vii) H2, 10% Pd/C, EtOH, (viii) LiAlH4, THS, rt, (ix) 1% TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy), NaClO, KBr, NaHCO3, DCM, H2O, (x) 2-(1,3-dioxolan-2-yl)methyltriphenylphosphonium bromide, t-BuOK, THF, rt. CHCOOEt, 150 °C, 2 h, (vii) H2, 10% Pd/C, EtOH, (viii) LiAlH4, THS, rt, (ix) 1% TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy), NaClO, KBr, NaHCO3, DCM, H2O, (x) 2-(1,3-dioxolan-2-yl)methyltriphenylphosphonium bromide, t-BuOK, THF, rt. | ||
In this study, a series of novel 13-pyridinealkyl berberine analogues (Scheme 1b) were synthesized and their DNA binding activity investigated by various biophysical experiments and molecular modeling studies. These analogues are named BER1, BER2, BER3, BER4 and BER5 based on the number of methylene groups in the linker chain. We observed the most efficient binding with BER5 bearing the highest number of CH2 groups in the series. Furthermore, these 13-pyridinealkyl berberine analogues induced apoptosis in HepG2 and MCF-7 human tumour cell lines. Reactive oxygen species (ROS) play pivotal roles in DNA damage16 and p53 plays a role as downstream effecter of ROS.17 The function of redox homeostasis in p53-regulated processes is a topic of great interest. Taken together, this work provides new fundamental knowledge and molecular mechanisms for the anticancer action of berberine analogues that may help further development as antineoplastic therapeutics.5,6,18,19
Results and discussion
Characterization of specific alkaloid–DNA association by spectroscopic analysis
These 13-pyridinealkyl berberine analogues possess an extra anchoring site. The binding of these analogues was initially probed by absorption spectroscopy titration methodology. The 13-pyridinealkyl analogues, like berberine, have characteristic absorption spectra in the region of 300–550 nm. The major peak maximam of the analogues varied around 340 nm and the spectral nature and features were similar. In this region DNA has negligible absorbance.In the absorption spectral titration, hypochromic and bathochromic effects were observed with the occurrence of isosbestic points for all the analogues on titration with DNA. The representative absorbance titration profile of BER5 with calf thymus (CT) DNA is depicted in Fig. 1A. The titration profiles of other analogues are presented in Fig. S1 (ESI†). The changes observed in the absorption spectra of BER5 are characteristic features of DNA intercalating ligands, revealing that the isoquinoline chromophore is stacked between the DNA base pairs in an intercalated posture. Equilibrium binding is confirmed from the presence of isosbestic points involving two state systems comprising of only free and bound alkaloid analogues. The alkaloid molecule along with the substituent moiety enters inside the DNA helix in such way that it is unable to form H-bonds with the solvent water molecules. The Scatchard plots of binding and subsequent McGhee–von Hippel analysis20 of the absorption data (Fig. S2, ESI†) revealed that the BER shows cooperative binding manifested by negative slope at low r values. These are described in details in the ESI.† On the other hand, the analogues present a non-cooperative binding mode with positive slope at low r values. So, the substitution of a pyridinealkyl group at the 13-position dramatically changed the nature of the binding from a non-cooperative mode to cooperative mode. This feature of these analogues is similar to that observed for 9 and 13-substituted analogues reported previously.18,19,21 The equilibrium binding affinity values and the number of excluded sites i.e., the number of base pairs excluded by the binding of an alkaloid as per neighbour exclusion model, on the binding of these analogues are depicted in Table S1 (ESI†). The highest binding constant was observed for BER5 with five CH2 groups in the linker and the affinity increased with increasing chain length except for BER1 where affinity was lower than BER. The number of excluded sites decreased with increasing chain length and was 6.6, 3.4, 3.2, 3.2 and 3.2, respectively, for BER1–5.
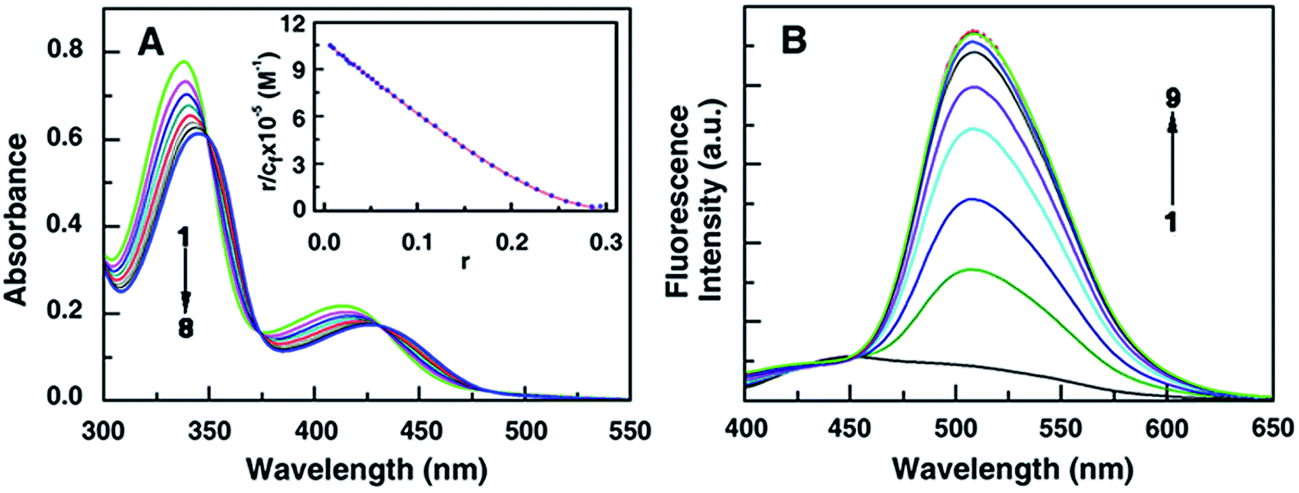 | ||
| Fig. 1 Absorbance spectral changes in BER5 (35 μM, curve 1) (A) with increasing concentration of DNA upto 400 μM. Inset: Scatchard plot for the binding of BER5 to DNA obtained from McGhee–von Hippel analysis of spectrophotometric data. (B) Fluorescence spectra of BER5 (5 μM, curve 1) with increasing concentration of DNA upto 70 μM of DNA. | ||
Berberine presents a weak intrinsic fluorescence property in aqueous buffers. The singlet excited state of the alkaloid returns to the ground state due to its intersystem crossing via nonradioactive mechanism.22 The 13-pyridinealkyl berberine analogues exhibited more intense fluorescence than BER. The fluorescence intensity enhanced remarkably on binding with DNA. A representative spectral titration in the case of BER5 is presented in Fig. 1B and the titration profiles of the analogues BER1–BER4 are presented in the Fig. S3 (ESI†). Change in the hydrophobic environment of the chromophore of the ligand due to transfer from a high polarity solvent environment to low polarity DNA interior when intercalated and the consequent electronic interactions appear to cause this enhanced fluorescence intensity. Furthermore, in the solution state the rotational movement of the free alkaloid analogue molecules favours a radiationless deactivation of the excited states. But when bound to DNA in the intercalative mode the rotation is hindered and the deactivation through fluorescence emission is favoured, and a significant increase in the fluorescence intensity occurs.
Conformational changes in DNA measured by circular dichroism spectroscopy
The right handed helical conformation of B-form DNA showed a characteristic CD spectrum with a positive band in the 275–280 nm region and a negative band around 248 nm (Fig. 2A). The long wavelength band arises due to the base stacking interactions and the 248 nm band is due to the helicity.23 The alkaloid analogues have no chiral center and hence are CD inactive. In an alkaloid–DNA titration study, the molar ellipticity value of the positive band increased with a small red shift of the wavelength maximum while the negative band changed only marginally. These features were similar in all the cases. The maximum change was observed in the case of BER5 as shown in the Fig. 2A. From the nature of the data it may be divulged that the alkaloid analogue molecules bind to DNA helix leading to conformational changes within in the B-form helical organization. It was also observed that a weak induced circular dichroism band appeared in the 340 nm region as shown in the Fig. 2B which is near the absorption maxima of the analogues. The result revealed that the alkaloid analogues bind in between the base pairs of the DNA helix by intercalation and due to this the helical chirality of DNA has changed. The induced CD spectrum arises from the strong interaction of the electric transition moments of the analogues and the transition moments of the chirally arranged DNA bases/base pairs.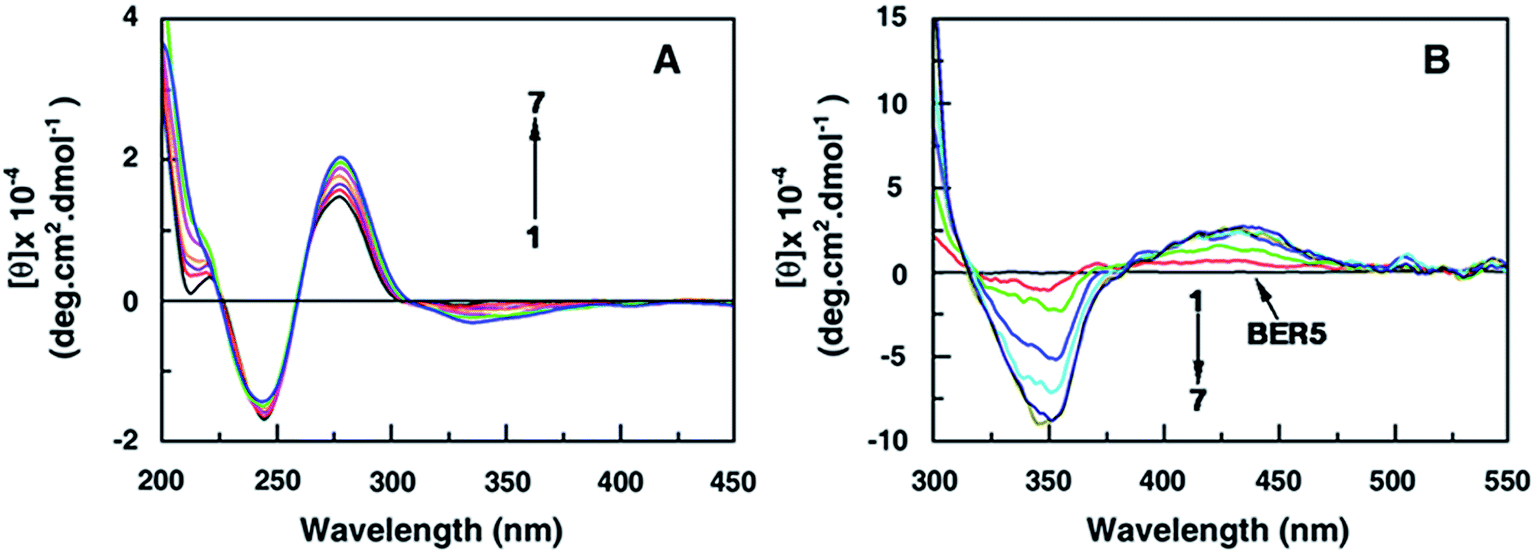 | ||
| Fig. 2 Circular dichroism spectra (A) of DNA (30 μM, curve 1) with increasing concentration of BER5 upto 30 μM. Induced CD spectra (B) of BER5 (60 μM, curve 1) with increasing concentration of DNA upto 150 μM as represented by curves 2–7. | ||
Thermal stabilization of the DNA helix by the alkaloids
Optical thermal melting experiments were carried out to understand the stabilization of the DNA helix on binding of the berberine analogues. The melting profile of DNA, monitored at the absorbance maximum (260 nm), revealed a cooperative hyperchromic transition of ∼40% with a melting temperature (Tm) around 65 °C under the conditions employed here. Berberine stabilized DNA by about 7 °C. The binding of BER5 induced the highest stabilization and the melting temperature was around 85 °C (ΔTm = 20 °C). With the exception of BER1 (ΔTm = 5 °C) all the analogues stabilized DNA more than BER. The results are presented in Table S2† and the corresponding melting profiles are shown in Fig. S4 (ESI†).The DNA stabilization was also confirmed by differential scanning calorimetry (DSC) experiments. The endothermic DNA peak was shifted to higher temperatures due to the binding of the alkaloid analogues. The ΔTm values were 8.00, 4.19, 13.33, 16.75, 17.94 and 19.57 °C, respectively, for BER and BER1 to 5. These values are close to those obtained from optical melting experiments. The results are depicted in Table S2 (ESI†).
Elucidation of the mode of binding of the alkaloid analogues by fluorescence quenching and viscosity methods
BER is known to bind to DNA in an intercalative mode.7,9,10 But to understand the mode of binding of the analogues we applied the fluorescence quenching technique and viscosity measurements. Fluorescence quenching using ferrocyanide ions, an anionic quencher, has been effectively used to distinguish between intercalation and groove-binding modes of berberine analogues18 and other small molecules.24–26The Stern–Volmer plots for fluorescence quenching of the BER and its analogues–DNA complexes by ferrocyanide ions are presented in Fig. S5 (ESI†). The results indicated that free analogue molecules were quenched efficiently by the ferrocyanide ions. More fluorescence quenching was observed in the case of BER1 than BER and the effect gradually decreased for bound BER2–5, indicating that the bound analogues BER2–5 are located in a relatively more protected environment inside the DNA helix compared to those unbound. The Stern–Volmer quenching constants (Ksv) calculated were in the range 215–230 M−1 for the unbound BER and analogues, and 95, 170, 80, 71, 39 and 33 M−1, respectively, for bound BER and BER1–5. The trend in the values revealed that the Ksv of BER1 was higher than that of BER and for the others it varied as BER > BER2 > BER3 > BER4 > BER5. Based on this data it can be inferred that BER1 is a weaker intercalator than BER, while BER2–BER5 are better intercalators than BER. The pyridyl group of BER1 and the isoquinoline chromophore are separated by a single (CH2) group and therefore the side chain linker appears to be too close to the isoquinoline chromophore leading to inhibition of the intercalation BER1. With gradual increase of the number of CH2 groups in the linker chain, intercalation capability was found to increase and it became maximum for BER5. This was also clearly evident from the viscosity data, where the helix length enhancement due to intercalation was maximum with BER5. A plot of relative increase in length of rod like DNA (L/Lo) versus r, where r is the number of berberine analogue molecules bound per mol of the DNA base pairs, is presented in Fig. S6 (ESI†). The slope of the plots (β values) for BER and BER1–BER5 binding to DNA was found to be 0.66, 0.56, 0.70, 0.76, 0.83 and 0.90, respectively, compared to value of 1.0 for classical intercalator. Taken together, these results confirmed the intercalation of these analogues to DNA and also suggested a better intercalation geometry for BER2–5 compared to BER and provided evidence that the linker chain with more than one CH2 group decisively contributes to the stronger intercalative binding of the isoquinoline moiety.
Thermodynamics of berberine analogues–DNA association
The thermodynamic profile of the interaction, that may provide key insights into the nature of the molecular forces that drive the complexation, was obtained from isothermal titration calorimetry (ITC) studies. Fig. 3A and B shows the representative ITC profiles of BER1 and BER5–DNA interaction. For BER5 the binding was exothermic in nature (Fig. 3A) but for BER1 (Fig. 3B) it showed endothermic heats. The ITC profiles in all the other cases were exothermic in nature. The presence of only one binding event in the thermogram in each case enabled the fitting of the isotherms to ‘one set of binding sites’ model yielding the best fit curve providing the equilibrium association constant (K), the stoichiometry (N) and the change in enthalpy (ΔrH0). The entropy contribution (TΔrS0) and the standard change in molar Gibbs energy (ΔrG0) were calculated as described previously.18 The data obtained from the ITC experiments are summarized in the Table 1. A comparative chart depicting the variation of the K values of the analogues are shown in Fig. 3C. In all the cases except BER1 the binding was favoured by negative enthalpy and positive entropy changes. In the case of BER1, the binding had a positive enthalpy term. The small positive enthalpy for the binding was unfavourable to the Gibbs energy. The large entropy contribution in the binding of BER1 compared to BER and other analogues can be interpreted to arise from the release of bound water and condensed sodium ions from the DNA grooves and phosphates, respectively. The pyridinealkyl moiety in BER1 appears to be very close to the isoquinoline chromophore of berberine leading to steric effects preventing BER1 from intercalating strongly between the DNA base pairs. This is also corroborated from the lower binding affinity of BER1 compared to all other analogues and even BER. A similar phenomenon was also reported previously for the DNA binding of 13-phenylalkyl berberine analogues.18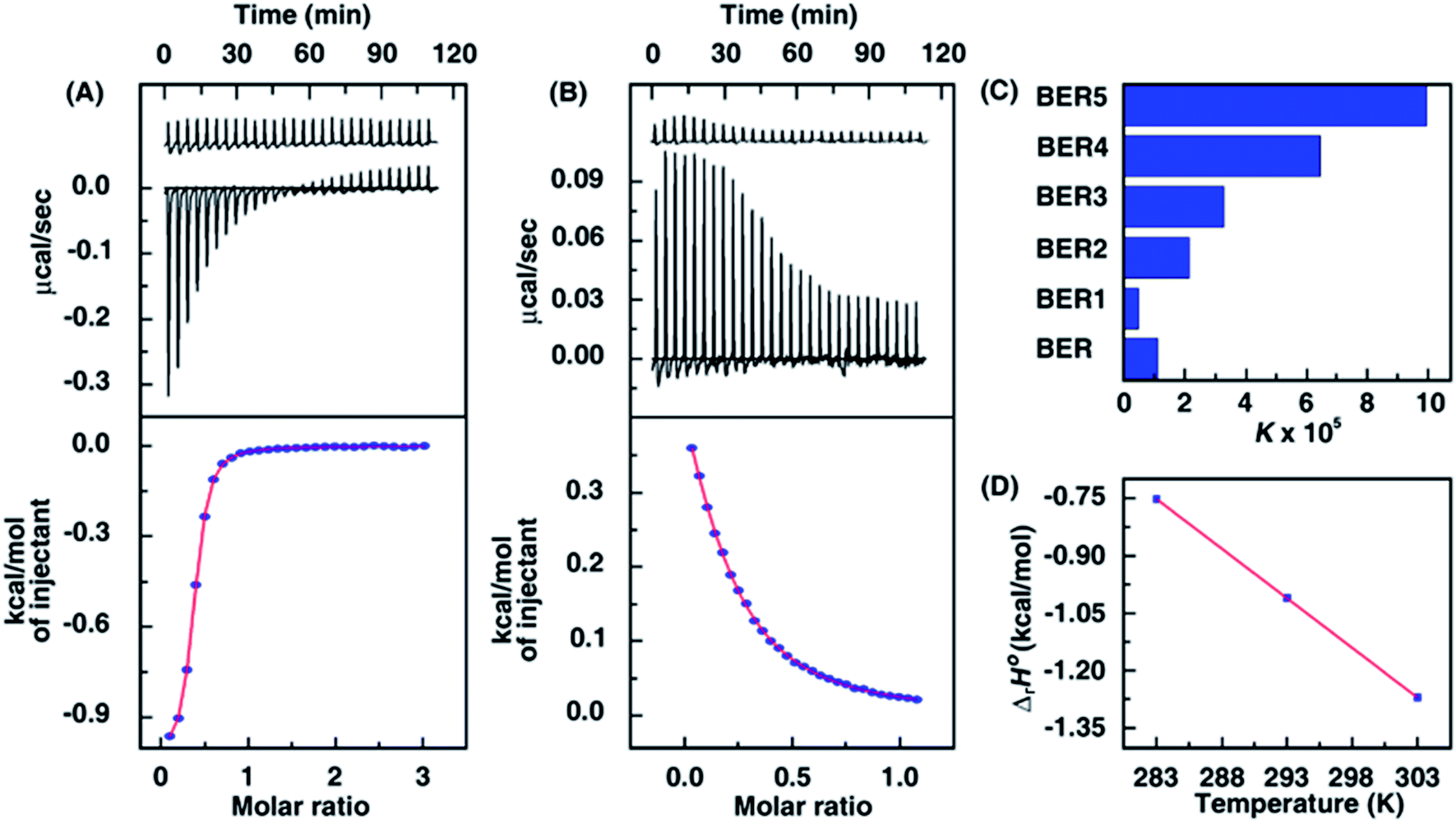 | ||
| Fig. 3 ITC profiles of BER5 (A) and BER1 (B) (500 μM) titrated into 50 μM and 100 μM, respectively, of DNA at 20 °C. The upper panels present the heat burst curves for the injection of samples and dilution profiles, and the bottom panels show the integrated heat data after correction of heat of dilution. The symbols represent the data points that were fitted to a model ‘one-site of binding sites’ and the solid lines represent the best-fit data. (C) Variation of the binding constants of BER and its analogues–DNA from ITC studies. (D) Variation of standard molar enthalpy change with temperature for BER5–DNA interaction. | ||
| Alkaloid analogue | Binding constant (K) × 105 M−1 | Stoichiometry (N) | (ΔrG0) kcal mol−1 | (ΔrH0) kcal mol−1 | (TΔrS0) kcal mol−1 |
|---|---|---|---|---|---|
a The ITC experiments were conducted at a pressure of 101.10 kPa at 20 °C. K, the binding constant, N, the binding stoichiometry and ΔrH0, the standard molar enthalpy change were evaluated from ITC profiles fitting to Origin 7.0 software using the ‘one set of binding sites’ model. The values of ΔrG0, standard molar Gibbs energy change and TΔrS0, the standard molar entropy contribution were determined using the equations ΔrG0 = ΔrH0 − TΔrS0 = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnK. R is the universal gas constant (1.98722 cal K−1 mol−1). lnK. R is the universal gas constant (1.98722 cal K−1 mol−1). |
|||||
| BER | 1.09 ± 0.07 | 0.482 ± 0.03 | −6.75 ± 0.53 | −2.834 ± 0.13 | 3.92 |
| BER1 | 0.46 ± 0.08 | 0.161 ± 0.08 | −6.25 ± 0.98 | 0.89 ± 0.18 | 7.14 |
| BER2 | 2.14 ± 0.05 | 0.471 ± 0.02 | −7.07 ± 0.96 | −1.80 ± 0.06 | 5.27 |
| BER3 | 3.27 ± 0.33 | 0.361 ± 0.07 | −7.39 ± 0.90 | −1.42 ± 0.09 | 5.97 |
| BER4 | 6.45 ± 0.08 | 0.356 ± 0.02 | 7.78 ± 0.61 | −1.190 ± 0.06 | 6.59 |
| BER5 | 9.96 ± 0.05 | 0.352 ± 0.05 | −8.04 ± 0.77 | −1.04 ± 0.09 | 7.00 |
Ionic strength and temperature dependence of the binding
As the berberine analogues are cationic in nature it is expected that ionic interactions might play a leading role in the complexation process. It was observed that the ionic strength of the medium had significant influence on the binding of BER analogues in many previously reported studies.18,27 To check on the nature of binding forces involved, the effect of ionic strength on the binding was investigated by ITC experiments in conjunction with van't Hoff analysis in the case of the strongest binding analogue, i.e., BER5 as a representative case. The thermodynamic parameters of the binding at three [Na+] concentrations investigated, viz. 10, 30 and 50 mM [Na+], were evaluated and the resulted are collated in Table S3 (ESI†). Increased [Na+] concentration results in reduced electrostatic repulsion between the negatively charged phosphate groups of adjacent nucleotides, and consequently the binding of BER5 to DNA was hindered, and the equilibrium constant of the complexation decreased; at 30 mM, the binding affinity was 4.6 × 105 M−1 which was about half of that at 10 mM [Na+].A relation linking the equilibrium constant with the Na+ ion concentration based on polyelectrolytic theories of Manning's counter ions condensation model28 has been derived as follows
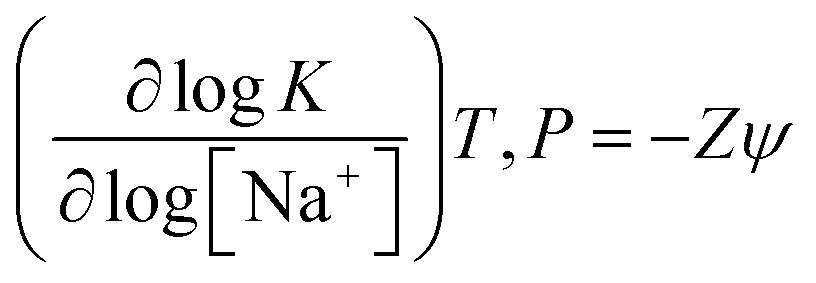
In this relation, Z is the apparent charge on the berberine analogue and ψ is the fraction of sodium ions bound per phosphate group of the DNA. The number of ions released on the binding of a single BER5 molecule to DNA was calculated to be 0.70 from the slope of the plot of logK versus log[Na+]. From the dependence of K on [Na+], the observed Gibbs energy was partitioned between the polyelectrolytic (ΔrGpe) and non-polyelectrolytic (ΔrGt) components using the relationship
| ΔrG0pe = ZψRTln([Na+]) |
Valuable insights into the type and magnitude of forces involved in the binding process was also obtained from the heat capacity (ΔrC0p) data derived from the temperature dependant variation of ΔrH0. Titration calorimetry experiments were performed at three different temperatures, viz. 10, 20 and 30 °C, respectively, for the binding of the analogue BER5. The thermodynamic parameters derived from these experiments are presented in Table S4 (ESI†). A plot of ΔrH0 versus T is presented in Fig. 3D. From the slope of the plot the value ΔrC0p was determined to be −25.97 cal mol−1 K−1 for BER5–DNA complexation. For berberine a value of −110 cal mol−1 K−1 was reported earlier.29 A negative heat capacity term is proposed to arise due to the removal of large amount of nonpolar surface from aqueous exposure upon complex formation resulting in dominant hydrophobic effects in the interaction and has been displayed by a number of small molecule binding to DNA and RNA. Deriving strength from these results it may be proposed that there is a strong hydrophobic element in the interaction of these berberine analogues with DNA.
Base pair specificity of BER5 from ITC experiments
Berberine is one of the few DNA intercalating molecules known to exhibit remarkable AT base pair specificity.9,10 To evaluate the binding specificity of the analogues we studied the binding of BER5, as a representative case, to poly(dA)·poly(dT) and poly(dG)·poly(dC). From ITC experiments the equilibrium constant for the binding to poly(dA)·poly(dT) and poly(dG)·poly(dC) were evaluated to be 2.26 × 106 M−1 and 4.96 × 105 M−1, respectively. It may be noted that the AT polynucleotide binding affinity was about 22 times higher than that to the GC polynucleotide. So it can be ascertained unequivocally that the AT base pair specificity of berberine is faithfully reproduced in these analogues also.Elucidation of the binding site of BER5 by molecular docking
Molecular docking studies provide some insight into the interaction between the drug and DNA, and the results may be correlated with the experimental data. Here we chosen two oligo nucleotides of different GC content of PDB identifier 258D (A) and 1JH1 (B). The BER5 molecule has two parts, one isoquinoline part (same as berberine) and an additional spacer arm of alkylpyridine part. In both cases the docking studies showed that the isoquinoline part is intercalated between the two base pairs of DNA and the additional alkylpyridine part formed H-bonding interactions with the base of the DNA molecule as shown in the Fig. 4. The distance between the H-bonding of N-atom of pyridine (BER5) and the C6–NH2 group of the adenine (DNA) is 2 Å for A and 2.3 Å for N-atom of pyridine and the C2–NH2 group of the guanine. The calculated binding energy values from theoretical studies due to binding with the oligomer were −8.59 and −7.90 kcal mol−1, respectively. The theoretical studies also supported our ITC data of base pair specificity of the BER5 molecule.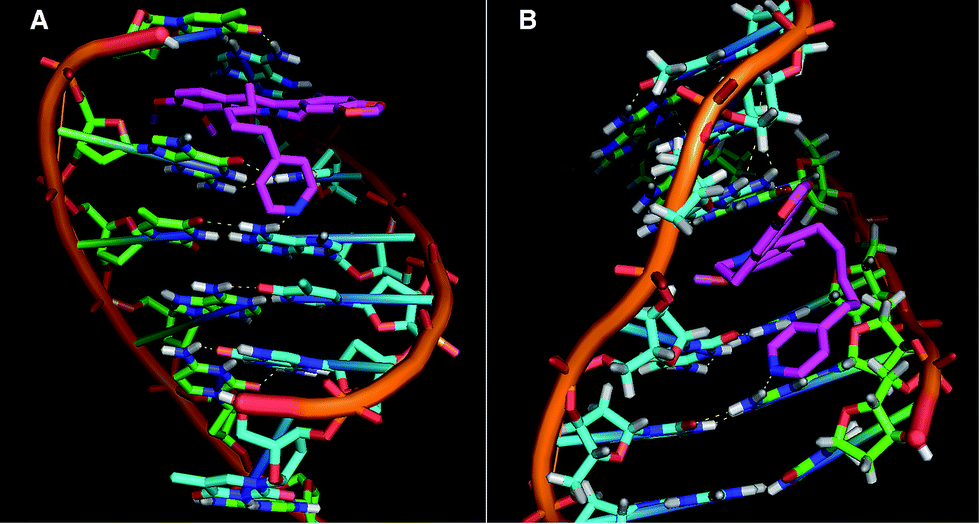 | ||
| Fig. 4 Two docked conformations (A and B) of BER5 with two different DNA fragments. | ||
Apoptotic effect of BER5 in HepG2 and MCF-7 human tumour cells
To evaluate the efficacy of the BER and its analogues against cancer, the compounds were tested for their cytotoxic effect against hepatocellular carcinoma (HepG2) and mammary human carcinoma (MCF-7) cell lines using the MTT assay. After 24 h treatment, it was found that among the five compounds, BER5 showed a higher degree of concentration-dependent increase in growth inhibition in HepG2 and MCF-7 cell lines, having GI50 values of 15.7 and 33.2 μM, respectively (Fig. 5A and B). However, it exhibited minimal effect on human embryonic kidney 293 (HEK-293) cell lines even at a concentration of 40 μM (Fig. 5C). Treatment with BER5 induced phosphatidylserine (PS) externalization in HepG2 and MCF-7 cells after 24 h treatment which is a hallmark of apoptosis. After 24 h treatment the percentage of apoptotic cells was 28.1% and 15.6% as compared to 5.3% and 2.0% in the vehicle treated cells in HepG2 and MCF-7 cell lines (Fig. 5D). Cell cycle arrest at various phases of cell division precedes apoptosis. Treatment of HepG2 and MCF-7 cells with BER5 (GI50 concentration) for 24 h showed a gradual increase in the number of cells in the G0/G1 phase. The percentages of G0/G1 population for the control and BER5 treated HepG2 cells were 57.3% and 67.5%, respectively, and for MCF-7 were 50.5% and 75.7%, respectively, indicating that BER5 induced cell cycle arrest at G0/G1 phase for these two cell lines (Fig. 5E). These results clearly suggested that BER5 induced apoptosis in HepG2 and MCF-7 cells.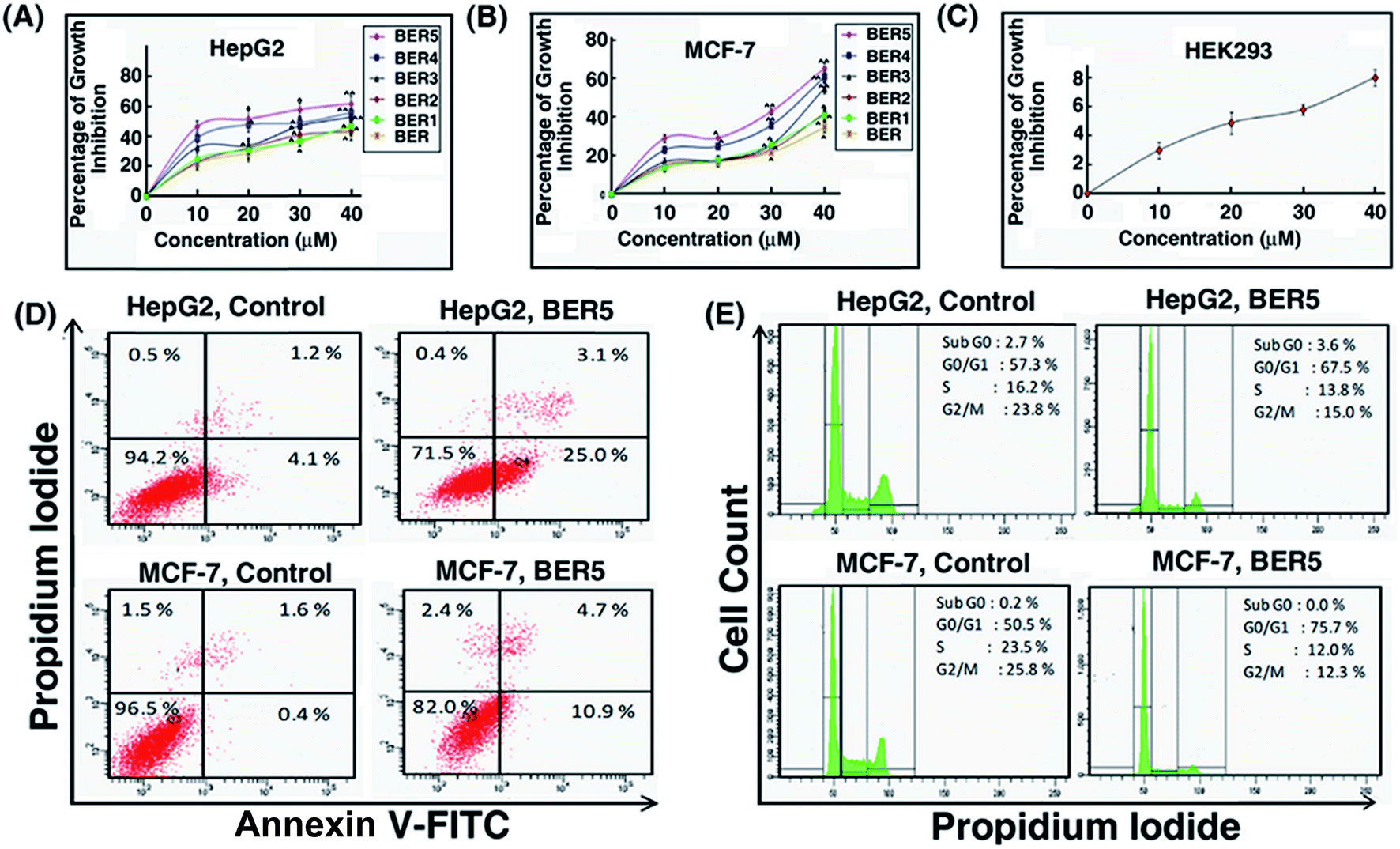 | ||
| Fig. 5 Percentage growth inhibition of HepG2 (A) and MCF-7 (B) following treatment with BER, BER1, BER2, BER3, BER4 and BER5 (0, 10, 20, 30 and 40 μM) for 24 h. (C) Growth inhibition of HEK293 cells following treatment with BER5 (0, 10, 20, 30 and 40 μM) for 24 h. (D) Flow cytometric analysis of apoptosis induction in HepG2 and MCF-7 cells after treatment with GI50 concentration of BER5 for 24 h. (E) Cell cycle analysis of BER5 (GI50) treated HepG2 and MCF-7 cells for 24 h. | ||
Effect of BER5 induced ROS generation on apoptosis
Reactive oxygen species (ROS) generation plays an important role in apoptosis, so it is necessary to investigate the ability of BER analogue to generate ROS. Cells were exposed to GI50 concentration of BER5 for 24 h and analyzed for the presence of ROS by flow cytometry. Generation of reactive oxygen species is a key factor of apoptotic cell death. A number of studies established that berberine induced ROS generation and this has been reviewed.2 To further elucidate whether BER5 induced intra cellular ROS generation in HepG2 and MCF-7 cells, ROS content was measured by flow cytometry with H2DCFDA. Treatment of BER5 at GI50 concentration with HepG2 and MCF-7 cells exhibited significantly enhanced intracellular ROS levels (Fig. 6A).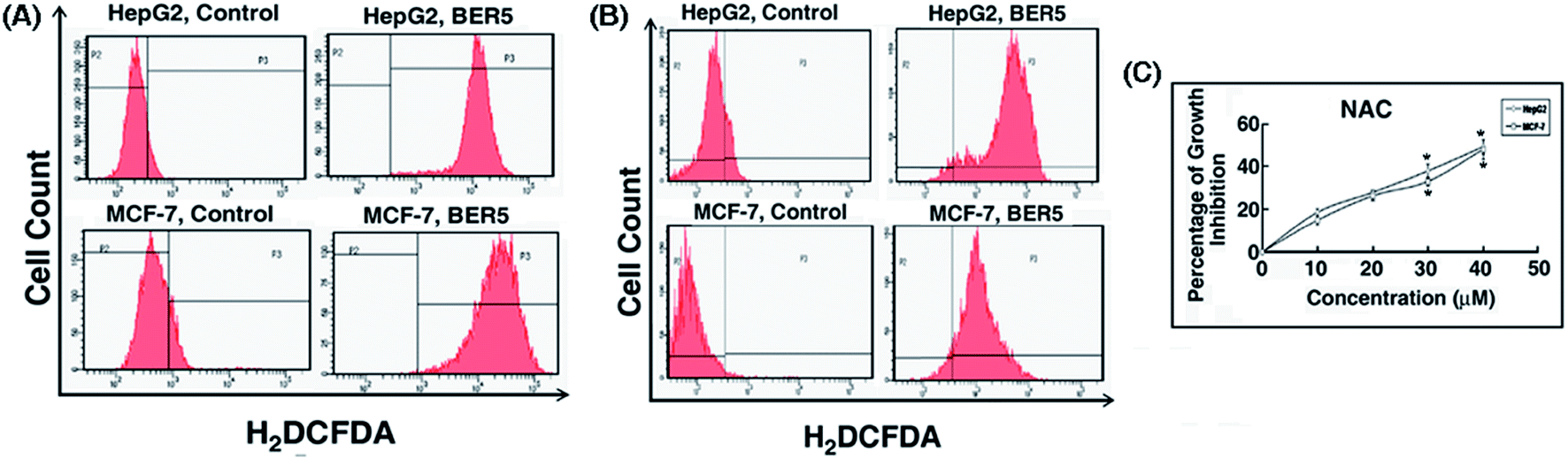 | ||
| Fig. 6 Analysis of ROS generation in HepG2 (A) and MCF-7 (B) cells following treatment with BER5 along with NAC after 24 h by flow cytometry. (C) Percentage of growth inhibition in HepG2 and MCF-7 cells in presence of either NAC. Values are mean ± S.D and represent one of the three representative experiments. *P < 0.05. | ||
Flow cytometric analysis revealed that the DCF fluorescence intensity in BER5 (GI50) treated cells was much higher for HepG2 and MCF-7 cells with respect to the control, indicating a sharp increase in mean fluorescence intensity from the control to BER5 treated cells. Upon treatment with GI50 concentration of BER5, there was an increase in intracellular ROS generation as compared with the control set after 24 h, which was dampened upon pre-incubating with NAC. Following pre-incubation with NAC, DCF fluorescence intensity of the control was lower with respect to BER5 treatment which was higher for HepG2 and MCF-7 cells, (Fig. 6B). Moreover, pre-incubation with NAC also inhibited the growth inhibitory role of BER5 (Fig. 6C). From this study it may be inferred that BER5 induced death in HepG2 and MCF-7 cells precede via the ROS mediated pathway.
Apoptotic cell death triggered by BER5 results in ROS dependent p53 activation
HepG2 and MCF-7 cells lines have wild type p53, which generally has a role in apoptosis. Therefore, the role p53 activation in BER5 induced apoptosis was examined.Treatment of HepG2 and MCF-7 cells with BER5 (GI50) resulted in increased expression and translocation of p53 to the nucleus along with increased expression of p21 as compared to the control (Fig. 7A). The corresponding fluorescence intensity graphs are presented in Fig. S7 (ESI†). Moreover, cells that were incubated with 20 μM of pifithrin-α (an inhibitor of p53 transactivation) for 2 h prior to treatment with BER5 showed inhibition in cytotoxicity by BER5, indicating that p53 may be involved in BER5 mediated apoptosis (Fig. 7C).
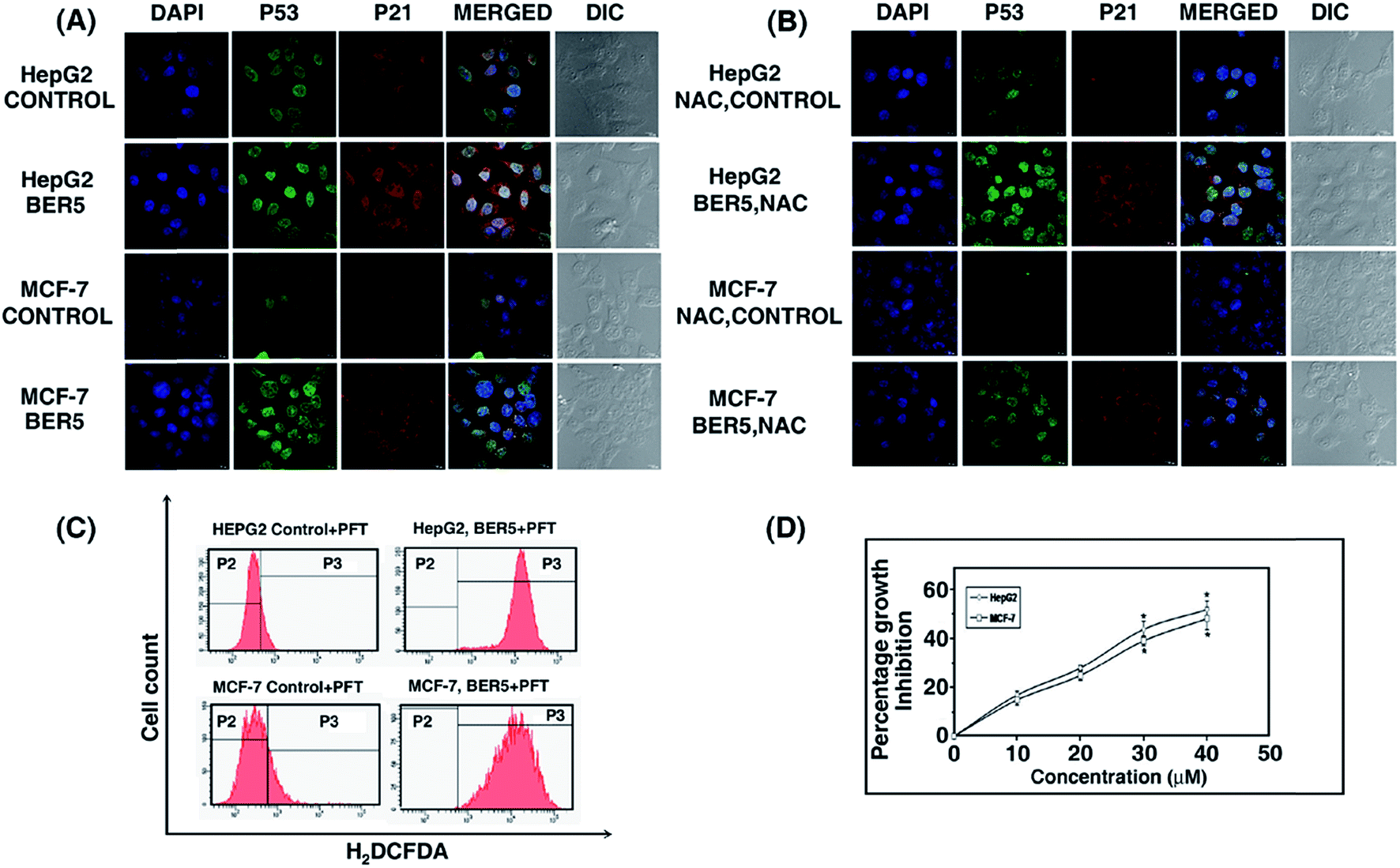 | ||
| Fig. 7 (A) Up regulation of p53, p21 and subsequent nuclear translocation of p53 upon treatment of cells with BER5 (0 and GI50 concentration). Scale bar = 15 mm. Magnification at 40×. (B) Down regulation of p53 and p21 in the presence of NAC. Scale bar = 15 mm. Magnification at 40×. (C) ROS generation of HepG2 and MCF-7 cells after treatment with BER5 (GI50) in the presence of PFT-α. (D) Percentage growth inhibition of HepG2 and MCF-7 cells in presence of pifithrin-α and BER5 with respect to the control (cells without treatment with BER5). | ||
In order to evaluate whether ROS is involved in p53 activation or vice versa, HepG2 and MCF-7 cells were pre-incubated with NAC prior to BER5 treatment, which led to the suppression of p53 and p21 expression (Fig. 7B) whereas, incubation with pifithrin-α, prior to BER5 exposure failed to inhibit ROS production (Fig. 7D), indicating that ROS is acting upstream of p53 but not vice versa. Thereby, it can be concluded that p53 induced apoptosis in these cell lines is ROS dependent.
Compound intake by BER5
From the biophysical studies presented above it was established that among the five alkaloid analogues BER5 showed the highest affinity to DNA. Therefore, it has become important to understand the cellular uptake and sub-cellular distribution of BER5 in order to gain further mechanistic insights. Previous reports established that BER has fluorescence property.30 BER5 exhibited fluorescence emission property and its fluorescence intensity was higher than BER and other analogues. From emission spectroscopy it was observed that upon binding to DNA the fluorescence intensity of BER5 increased (Fig. 1B). In order to visualize BER5 directly in the HepG2 and MCF-7 cells, cells were grown onto a cover slip and treated with BER5 as described in the Experimental section, and was evaluated using a confocal microscope at 12 h and 24 h time periods. DAPI was used as the nuclear marker. The fluorescence intensity of the analogue in cells was evaluated at 12 h and 24 h and it was found that at 12 h dense fluorescence intensity around nucleus (Fig. 8A) and at 24 h fluorescence was detected inside the nucleus (Fig. 8B) which proved that with time BER5 enters the cell nucleus. The uptake of BER5 was measured by treating cells for 24 h with BER5 (GI50) and measuring using flow cytometry which indicated a higher fluorescence in both HepG2 and MCF-7 cells as compared to the control (Fig. 8C).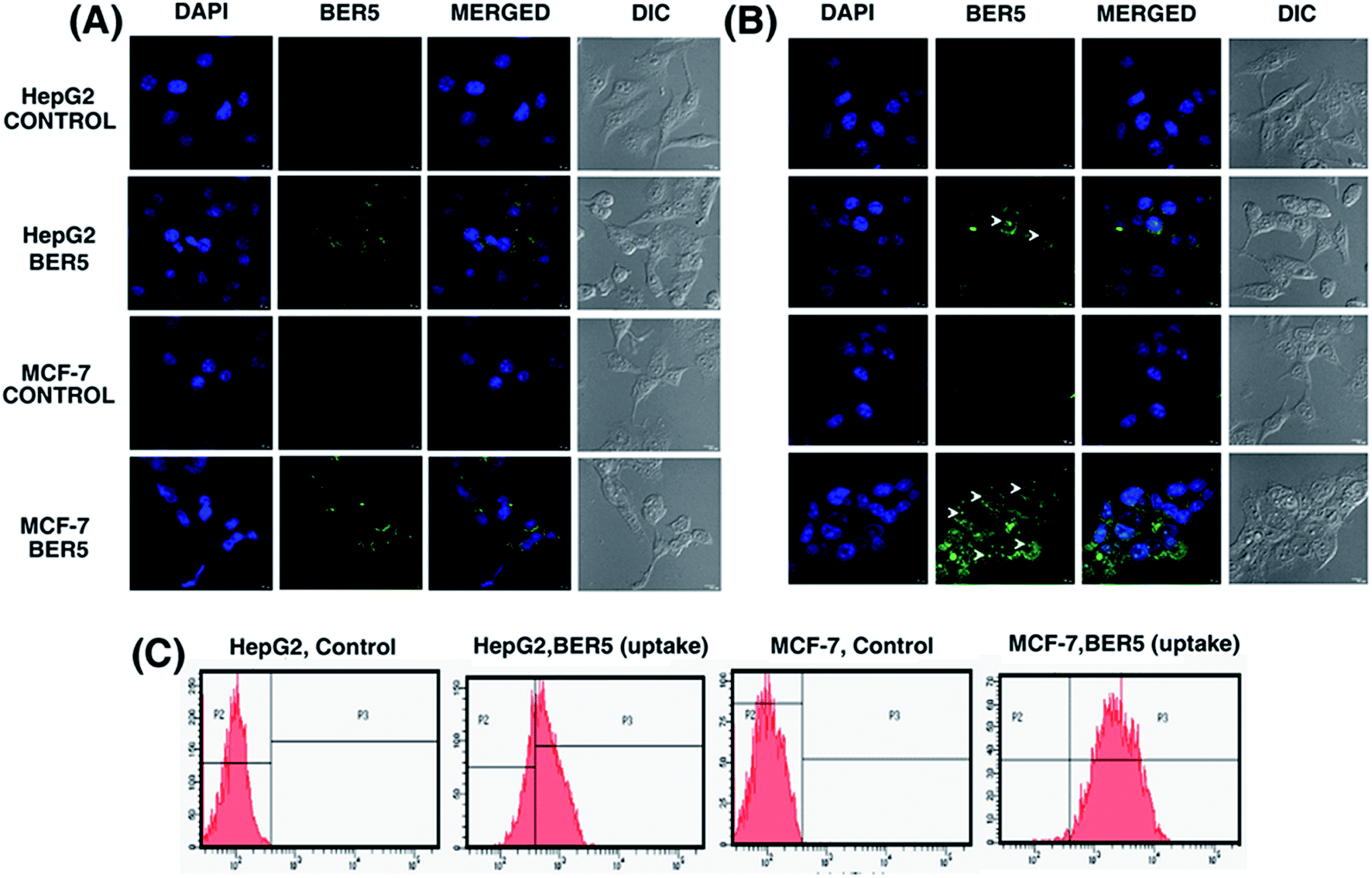 | ||
| Fig. 8 Confocal microscopic images showed BER5 (GI50) uptake at 12 h (A) and 24 h (B). (C) Flow cytometric analysis showed BER5 (GI50) uptake after 24 h. | ||
Conclusions
Here we have presented results on the DNA interaction of five new berberine analogues with pyridinealkyl substitution at the 13-position of the isoquinoline chromophore. The results obtained from absorption and fluorescence studies suggested at first that the analogues bound to DNA stronger than berberine, and that the increased affinity is applicable only when the linker chain length was beyond one CH2 group. Thus, a critical length was required for effecting higher binding. The binding affinity of the analogue BER5 was almost ten times higher than that of berberine. Unlike berberine, the analogues bound DNA in a non-cooperative manner and stabilized the DNA helix strongly against thermal strand separation. Similar results have been reported previously for analogues with substituents at 9- and 13-positions.18,21 The binding of these analogues have been inferred to occur through intercalation based on the results from viscosity and fluorescence quenching experiments, and further confirmed from molecular modeling studies. The conformational changes effected in DNA on binding and the induced circular dichroism acquired by the analogues further substantiated their stronger binding over berberine. From calorimetry results it was revealed that the analogues bound to DNA by an entropy driven manner that is justified because of their long side chain that can effect strong disruption of the water structure in the grooves of the DNA. Furthermore, the results of salt and temperature dependent studies proposed that the binding mostly involved non-polyelectrolytic forces which were corroborated from the value of the heat capacity change. Overall, the structural studies present a remarkably stronger affinity and better interaction profile for these analogues over berberine to DNA.Among the five 13-pyridinealkyl berberine analogues, BER5 showed maximum cytotoxicity towards HepG2 and MCF-7 human tumour cells after 24 h, but in different degrees. Interestingly, 40 μM of BER5 did not show any toxic effects on normal HEK293 cells suggesting a potential therapeutic window. Significant annexin V-FITC positivity and accumulation of cells at G0/G1 phase had indicated that BER5 induced apoptosis in HepG2 and MCF-7 cells.
As HepG2 and MCF-7 cells are p53 wild type, the role of p53 is quite relevant here. Pre-incubation with NAC resulted in an increase in cell viability in the presence of BER5 while pre-incubation with PFT-α did not prevent ROS generation. However, p21 the transcriptional targets of p53 was affected by the inhibitory role of NAC on ROS. BER5, therefore, may be inducing ROS generation upstream of p53 leading to apoptosis in the ROS mediated pathway. To conclude, BER5 mediates apoptosis in HepG2 and MCF-7 cells via a ROS mediated p53 dependent pathway and makes it a promising anticancer agent towards p53 wild type tumors.
Materials and methods
Materials
DNA from calf thymus (CT, type XI, 42% guanine–cytosine composition), poly(dA)·poly(dT), poly(dG)·poly(dC), berberine chloride (BER), L-glutamine, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), fetal bovine serum (FBS), streptomycin, penicillin, Dulbecco's modified eagle medium (DMEM), 4-2-hydroxyethyl-1-piperazineethanesulfonic acid (HEPES), JC-1, 6-diamidino-2-phenylindole dihydrochloride (DAPI), PI, annexin V-FITC, and other general reagents were purchased from Sigma (St. Louis, USA). Antibodies against p53 and p21 were purchased from Cell Signalling (Danvers, USA). The biophysical experiments were performed in citrate–phosphate (CP) buffer of pH 7.4, containing 10 mM Na2HPO4. Concentration was measured in a spectrophotometer by using molar absorption coefficients (ε, in M−1 cm−1) as follows. Calf thymus DNA: 13.2 × 102 (at 260 nm),18 poly(dA)·poly(dT): 13.6 × 102 at 260 nm, poly(dG)·poly(dC): 16.8 × 102 at 255 nm, berberine: 22.5 × 102 (at 345 nm)19 and all the 13-analogues: 21.3 × 102 at their respective λmax.Synthesis of 13-pyridinealkyl berberine derivatives BER1–BER5
The 13-pyridinelalkyl berberine derivatives were claimed in US Pat. 8,188,109B2 to Naxospharma srl. These were synthesized starting from commercial sample of berberine chloride hydrate (ca. 17% H2O, Shanghai Trust & We, China) and the appropriate pyridine (alkyl)carboxaldehydes (Fig. 1a), via a modification of a unusual enamine–aldehyde condensation performed on dihydroberberine31 and purified by following the method of Weber.32 The aldehydes were prepared starting from commercial pyridine-4-carboxaldehyde (Scheme 1b) or 4-(2-hydroxyethyl)pyridine (Scheme 1c) (both purchased from Alfa Aesar GmbH & Co KG, Karlsruhe, Germany), following usual homologation and oxidation methods and procedures known in organic chemistry.The purity (>94%) of the derivatives was ascertained on the Jasco HPLC LC-2000 series system (Jasco International, Europe) with an Agilent Eclipse XDB-C18 (4.6 mm × 150 mm × 3.5 mm) column (Agilent Technologies, USA) by applying a method by Weber.32 The rate of flow of the mobile phase (68% 30 mM ammonium acetate, 14 mM TEA, pH ∼ 4.85, 32% acetonitrile) was maintained at 1 mL min−1 and absorbance was measured at four wavelengths viz. 235, 265, 340, and 420 nm, respectively.
The structures of the derivatives were confirmed by 1H NMR spectra measured on a Varian Mercury 400 MHz spectrometer.
Methods
The following equation was used to evaluate ΔrG0.
| ΔrG0 = −RTlnK |
The binding entropy contribution (TΔrS0) was evaluated by the relationship
| TΔrS0 = ΔrH0 − ΔrG0 |
In these equations R and T represent the universal gas constant and absolute temperature in Kelvin, respectively.
Acknowledgements
This work was funded by grants from the Council of Scientific and Industrial Research (CSIR), Govt of India net work project BSC0123 (GenCODE). S. Chatterjee is a Senior Research Fellow supported by the University Grants Commission, Govt of India. Authors express sincere thanks to Dr S. N. Bhattacharyya, Principal Scientist and Mr D. Sarkar of this institute for help during confocal microscopic analysis. Ms A. Ganguly's assistance in flow cytometric experiments is appreciated. Naxospharma srl, Italy, acknowledges financial support by the Italian Ministry of Economic Development (Grant No. 01705) under the 6th call of the EuroTransBio initiative, transnational project BER.T.A (BERberine as antiTumour Agents).References
- C. M. Tsang, Y. C. Cheung, V. W. Y. Lui, Y. L. Yip, G. Zhang, V. W. Lin, K. C. P. Cheung, Y. Feng and S. W. Tsao, BMC Cancer, 2013, 13, 619 CrossRef PubMed.
- Y. Sun, K. Xun, Y. Wang and X. Chen, Anti-Cancer Drugs, 2009, 20, 757–769 CrossRef CAS PubMed.
- S. Jantová, L. Cipák, M. Cernáková and D. Kost'álová, J. Pharm. Pharmacol., 2003, 55, 1143–1149 CrossRef PubMed.
- J. P. Lin, J. S. Yang, J. H. Lee, W. T. Hsieh and J. G. Chung, World J. Gastroenterol., 2006, 12, 21–28 CAS.
- L. M. G. Ortiz, P. Lombardi, M. Tillhon and A. I. Scovassi, Molecules, 2014, 19, 12349–12367 CrossRef PubMed.
- L. M. G. Ortiz, M. Tillhon, M. Parks, I. Dutto, E. Prosperi, M. Savio, A. G. Arcamone, F. Buzzetti, P. Lombardi and A. I. Scovassi, BioMed Res. Int., 2014, 924585 Search PubMed.
- M. Ferraroni, C. Bazzicalupi, A. R. Bilia and P. Gratteri, Chem. Commun., 2011, 47, 4917–4919 RSC.
- M. Maiti and G. Suresh Kumar, J. Nucleic Acids, 2010, 593408 Search PubMed , 23 pages.
- K. Bhadra and G. Suresh Kumar, Med. Res. Rev., 2011, 31, 821–862 CrossRef CAS PubMed.
- D. Debnath, G. Suresh Kumar, R. Nandi and M. Maiti, Indian J. Biochem. Biophys., 1989, 26, 201–208 CAS.
- M. Halimani, S. P. Chandran, S. Kashyap, V. M. Jadhav, B. L. V. Prasad, S. Hotha and S. Maiti, Langmuir, 2009, 25, 2339–2347 CrossRef CAS PubMed.
- S. Hazra, M. Hossain and G. Suresh Kumar, J. Inclusion Phenom. Macrocyclic Chem., 2014, 78, 311–323 CrossRef CAS.
- S. P. Liu, Z. Yang, Z. F. Liu, J. T. Liu and Y. Shi, Anal. Chim. Acta, 2006, 572, 283–289 CrossRef CAS PubMed.
- E. Pierpaoli, V. Viola, A. Barucca, F. Orlando, F. Galli and M. Provinciali, Carcinogenesis, 2013, 34, 1352–1360 CrossRef CAS PubMed.
- E. Pierpaoli, E. Damiani, F. Orlando, G. Lucarini, B. Bartozzi, P. Lombardi, C. Salvatore, C. Geroni, A. Donati and M. Provinciali, Carcinogenesis, 2015, 36, 1169–1179 CrossRef PubMed.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS PubMed.
- B. Liu, Y. Chen and D. K. St Clair, Free Radicals Biol. Med., 2008, 44, 1529–1535 CrossRef CAS PubMed.
- D. Bhowmik, M. Hossain, F. Buzzetti, R. D'Auria, P. Lombardi and G. Suresh Kumar, J. Phys. Chem. B, 2012, 116, 2314–2324 CrossRef CAS PubMed.
- D. Bhowmik, F. Buzzetti, G. Fiorillo, F. Orzi, T. M. Syeda, P. Lombardi and G. Suresh Kumar, MedChemComm, 2014, 5, 226–231 RSC.
- J. D. McGhee and P. H. von Hippel, J. Mol. Biol., 1974, 86, 469–489 CrossRef CAS.
- A. Basu, P. Jaisankar and G. Suresh Kumar, Bioorg. Med. Chem., 2012, 20, 2498–2505 CrossRef CAS PubMed.
- K. Hirakawa, S. Kawanishi and T. Hirano, Chem. Res. Toxicol., 2005, 18, 1545–1552 CrossRef CAS PubMed.
- M. Rahban, A. Divsalar, A. A. Saboury and A. Golestani, J. Phys. Chem. B, 2010, 114, 5798–5803 CAS.
- M. M. Islam, P. Pandya, S. Roy Chowdhury, S. Kumar and G. Suresh Kumar, J. Mol. Struct., 2008, 891, 498–507 CrossRef CAS PubMed.
- S. Roy Chowdhury, M. M. Islam and G. Suresh Kumar, Mol. BioSyst., 2010, 6, 1265–1276 RSC.
- A. Das and G. Suresh Kumar, Biochim. Biophys. Acta, 2013, 1830, 4708–4718 CrossRef CAS PubMed.
- D. Bhowmik, F. Buzzetti, G. Fiorillo, L. Franchini, T. M. Syeda, P. Lombardi and G. Suresh Kumar, J. Therm. Anal. Calorim., 2014, 118, 461–473 CrossRef CAS.
- M. T. Record Jr, C. F. Anderson and T. M. Lohman, Q. Rev. Biophys., 1978, 11, 103–178 CrossRef CAS.
- K. Bhadra, M. Maiti and G. Suresh Kumar, Biochim. Biophys. Acta, 2008, 1780, 1054–1061 CrossRef CAS PubMed.
- M. M. Islam, S. Roy Chowdhury and G. Suresh Kumar, J. Phys. Chem. B, 2009, 113, 1210–1224 CrossRef CAS PubMed.
- K. Iwasa, M. Kamigauchi, M. Sugiura and H. Nanba, Planta Med., 1997, 63, 196–198 CrossRef CAS PubMed.
- H. A. Weber, M. K. Zart, A. E. Hodges, H. M. Molloy, B. M. O'Brien, L. A. Moody, A. P. Clark, R. K. Harris, J. D. Overstreet and C. S. Smith, J. Agric. Food Chem., 2003, 51, 7352–7358 CrossRef CAS PubMed.
- J. B. Chaires, Biochemistry, 1983, 22, 4204–4211 CrossRef CAS.
- K. Bhadra, M. Maiti and G. Suresh Kumar, Biochim. Biophys. Acta, 2007, 1770, 1071–1080 CrossRef CAS PubMed.
- M. M. Islam, P. Pandya, S. Kumar and G. Suresh Kumar, Mol. BioSyst., 2009, 5, 244–254 RSC.
- K. Bhadra, M. Maiti and G. Suresh Kumar, Chem. Biodiversity, 2009, 6, 1323–1342 CAS.
- M. M. Islam, R. Sinha and G. Suresh Kumar, Biophys. Chem., 2007, 125, 508–520 CrossRef CAS PubMed.
- R. Sinha, M. M. Islam, K. Bhadra, G. Suresh Kumar, A. Banerjee and M. Maiti, Bioorg. Med. Chem., 2006, 14, 800–814 CrossRef CAS PubMed.
- M. Hossain and G. Suresh Kumar, J. Chem. Thermodyn., 2010, 42, 1273–1280 CrossRef CAS PubMed.
- S. Mallick, P. Ghosh, S. K. Samanta, S. Kinra, B. C. Pal, A. Gomes and J. R. Vedasiromoni, Cancer Chemother. Pharmacol., 2010, 66, 709–719 CrossRef CAS PubMed.
- A. Nandy, S. K. Dey, S. Das, R. N. Munda, J. Dinda and K. Das Saha, Mol. Cancer, 2014, 13, 57 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra17214d |
| This journal is © The Royal Society of Chemistry 2015 |