
Carbon monoxide interactions with pure and doped B11XN12 (X = Mg, Ge, Ga) nano-clusters: a theoretical study
Abstract
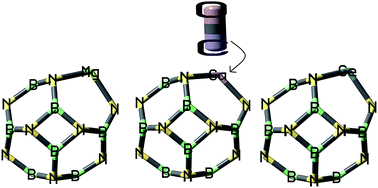
The goal of this investigation was to study a novel sensor for detecting the toxic gas compounds of CO using B11XN12 (X = Ge, Mg, and Ga) nano-clusters in terms of its energetic, geometric, and electronic structure using DFT calculations by the PBE-D method. The reaction of CO gas with these doping atoms results in a weak interaction and an elongation of X–N bond of B11XN12 nano-clusters. After the adsorption of CO gas over the doped positions of B11XN12 nano-cluster, the conductivity of the adsorbent and the atomic charges in some of the nearby B and N atoms around X atoms were dramatically enhanced. These calculations represent the capabilities of the B11XN12 nano-clusters in designing novel materials based on B11XN12 for potential applications in gas sensing.
 Please wait while we load your content...
Please wait while we load your content...

