
Fluorous bispidine: a bifunctional reagent for copper-catalyzed oxidation and knoevenagel condensation reactions in water†
Wei Jie Ang,
Yong Sheng Chng and
Yulin Lam*
Department of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543, Singapore. E-mail: chmlamyl@nus.edu.sg
First published on 17th September 2015
Abstract
Fluorous bispidine-type ligands have been developed to facilitate its recovery and reusability and to demonstrate its bifunctional property as a ligand and base in copper-catalyzed aerobic oxidation, the Knoevenagel condensation and tandem oxidation/Knoevenagel condensation in water under mild conditions. Application of the fluorous ligand was also extended to the surfactant-free copper-catalyzed allylic and benzylic sp3 C–H oxidation reaction in water. The fluorous ligands could be recovered using F-SPE with recovery ranging from 91–97% and could be reused five times with little loss of activity.
Introduction
Enzymes containing heme, non-heme iron or copper active sites play important roles in a large number of oxidative transformations and catalysis in biological systems.1 Increased knowledge of these enzymes and their biological functions has allowed the design and development of chemically similar biomimetic models. One such example is the iron–bispidine complex which is extensively used in oxidation catalysis.2Besides iron, copper, because of its natural abundance and redox potential, is another metal utilized in nature for many biological oxidations.3 Despite the economic and environmental benefits of copper-based catalysts, bispidine–copper complexes have thus far been applied mainly to aziridination.4 To our knowledge, the sole oxidation using bispidine–copper complex is the conversion of catechol in O2 to quinone.5 Our interest in developing recyclable catalyst/reagent for greener procedures6 thus led us to synthesize fluorous bispidine compounds (Fig. 1) and explore their applications as ligands for copper-catalyzed aerobic oxidation of benzylic alcohols and as a bifunctional reagent in the tandem oxidation/Knoevenagel condensation reaction.
 | ||
| Fig. 1 Bispidine-type ligands (L1–L3) and their fluorous analogous (FL1–FL3). | ||
Oxidation of benzylic alcohols is an important transformation as the corresponding benzylic aldehydes are versatile intermediates with a wide range of utility for perfumery, drugs and synthesis of fine chemicals.7 Classically, such oxidations are achieved with stoichiometric amounts of oxidants such as permanganates, chromium reagents, the Dess–Martin periodinane or IBX8 which present significant environmental issues with their use as huge amounts of waste are generated. The Cu-based, catalytic alcohol oxidations methods by Stahl, Sheldon and others were alternatives to the classical oxidation methods.9 Oxidation with greener alternative, such as O2 or air in water using the Cu salt/TEMPO system has received high interest.9g–m However these reactions generally require heating,9g,h,j,l use of pressurized oxygen9l and/or addition of base9g–j,l,m or surfactant.9i These reaction conditions together with the inability to recover the catalyst decreases the “greenness” of the procedure. Hence if the virtues of copper could be leveraged by its utilization at ambient temperature with water as a solvent and air as an oxidant, a greener and more robust protocol would result.
Besides aerobic oxidation, the allylic and benzylic sp3 C–H oxidation is another important organic transformation to generate α,β-unsaturated enones and benzylic ketones which are important building blocks in chemical syntheses and pharmacophores intermediates.10 To-date several protocols for sp3 C–H oxidation catalyzed by metal complexes in combination with tert-butyl hydroperoxide (TBHP) have been reported.11 However amongst the different metals, fewer oxidations have been carried out using Cu as catalyst. Previously, we reported the allylic and benzylic sp3 C–H oxidation using T-Hydro (70% TBHP in H2O) in water and SDS as a surfactant.6c However, the possibility of a surfactant-free protocol would increase the green aspect of the reaction condition. Thus, we herein expanded our investigation on the general usefulness of fluorous bispidine ligands to surfactant-free allylic and benzylic sp3 C–H oxidation reaction in water.
Since the nitrogens on bispidine are basic,12 we hypothesized that it could be used as a proton sponge and evaluated our fluorous bispidine compounds for its catalytic activity as a potential proton sponge in the Knoevenagel condensation of aldehydes and ketones with malononitrile in water. In this condensation reaction, bases such as NaOH, NaOEt and piperidine in organic solvents are commonly employed13 and the inability to recycle the base results in the production of large amounts of waste and corrosion which are detrimental to the environment. Various environmentally benign solid bases14 have been developed in recent years and the use of water as solvent has also greatly increased the environmentally friendliness of the reaction.
The efficiency and greenest of a synthetic sequence could be further enhanced by carrying out two or more chemical transformations in a ‘one-pot’ manner without the need for intermittent work-up and purification. The efficiency of these reactions is also greatly increased when the same catalyst is employed in each step. With water as the sole by-product in the bispidine–copper catalyzed benzylic alcohol oxidation reaction, this procedure allows the in situ formation of aldehydes which can be used, without purification, in a subsequent reaction in a tandem manner. Thus we were interested to explore if we could extend the bispidine–copper catalyzed benzylic alcohol oxidation reaction to a tandem process. The dual function of our fluorous bispidine-type ligand would allow the combination of the two separate reactions into a tandem oxidation/Knoevenagel condensation reaction which provides the possibility to save effort, time and resources in a manifold manner. To our knowledge, a tandem benzylic alcohol oxidation/Knoevenagel condensation in aqueous media using bimetallic Au–Pd catalyst was reported earlier.15a Heating (80 °C) was required for both steps of the reaction and molecular oxygen was used as an oxidant in the oxidation step. We herein report a tandem procedure using earth abundant copper metal as catalyst and air as an oxidant at milder conditions (ambient temperature to mild heating of 40 °C).
Results and discussion
Synthesis of ligands
Ligand L1 was prepared from piperidone 1 (ref. 16) via a Mannich reaction with 4-methoxybenzylamine and formaldehyde in refluxing ethanol in 80% yield and ligand L2 was synthesised according to reported procedure17 and obtained in 63% yield (Scheme 1). Fluorous aldehyde p2 was prepared according to reported procedure6c and was reduced using sodium borohydride (NaBH4) in THF to obtain the fluorous benzyl alcohol p2′ in quantitative yields. Chlorination of the alcohol p2′ using thionyl chloride (SOCl2) provided the fluorous benzyl chloride 2 in quantitative yield. Fluorous version of L1, FL1, was obtained from the alkylation of compound 3 (ref. 18) with fluorous benzyl chloride 2 in 85% yield. Analogous to FL1, FL2 was prepared by the alkylation of compound 3 and 1-iodo-1H,1H,2H,2H-perfluorodecane (C8F17CH2CH2I) in refluxing acetonitrile in 59% yield. Ligands L3 and FL3 were prepared by the reduction of bispidone 4 (ref. 19) with NaBH4 to give the bispidole 5 (94% yield) which in turn was treated with sodium hydride (NaH) and methyl iodide (CH3I) or C8F17CH2CH2I to produce L3 (83% yield) or FL3 (59% yield) respectively. | ||
| Scheme 1 Synthesis of bispidine-type ligands (L1–L3) and their fluorous analogous (FL1–FL3). | ||
Aerobic oxidation
For the initial assessment of the aerobic oxidation of benzylic alcohols, 4-methoxybenzyl alcohol 6a was chosen as the model substrate with 5 mol% CuBr, 5 mol% L1 as ligand, 5 mol% TEMPO, 10 mol% K2CO3 and water as the solvent under ambient air (open vessel) at room temperature. The desired 4-methoxybenzaldehyde 7a was obtained after 12 h in 41% yield (Table 1, entry 1). Gratifyingly, no overoxidized product (carboxylic acid) was observed. Due to the basic nature of bispidine-type ligands, we attempted the reaction in the absence of the base (K2CO3) and unexpectedly, the yield of compound 7a increased to 51% (Table 1, entry 2). To optimize the reaction, we varied the metal catalyst and found that the reaction was most efficient with CuBr in aqueous medium (Table 1, entries 3–8). Subsequently, we screened various flourous ligands and established that the reaction was most efficient with FL2 (Table 1, entries 9–11).
|
|||
|---|---|---|---|
| Entry | Cu salt | Ligand | Yieldb (%) |
| a Reaction condition: 4-methoxybenzyl alcohol 6a (0.5 mmol), Cu salt (5 mol%), ligand (5 mol%), TEMPO (5 mol%), ambient air, water (0.5 mL), r.t.b Isolated yields. Purity is >95% (according to NMR).c 10 mol% K2CO3. | |||
| 1c | CuBr | L1 | 41 |
| 2 | CuBr | L1 | 51 |
| 3 | CuBr2 | L1 | 38 |
| 4 | CuI | L1 | 7 |
| 5 | Cu(OAc)2·H2O | L1 | 38 |
| 6 | CuSO4·5H2O | L1 | 40 |
| 7 | Cu(BF4)2·H2O | L1 | 47 |
| 8 | Cu(ClO4)2·6H2O | L1 | 24 |
| 9 | CuBr | FL1 | 87 |
| 10 | CuBr | FL2 | 90 |
| 11 | CuBr | FL3 | 9 |

We also examined the possibility of using iron catalyst but this provided compound 7a in much lower yield (Table 2, entries 2–5). Control experiments were also carried out and in the absence of TEMPO and air, little or no product was observed (Table 2, entries 6 and 7). In the absence of FL2, only 57% of the desired product was obtained with 13% of the starting material recovered (Table 2, entry 8). We also examined the effect of varying the ratio of CuBr, FL2 and TEMPO but these changes provided compound 7a in lower yields (Table 2, entries 9–12). Finally, we determined the minimum time for the reaction to reach completion at room temperature to be 5 h (Table 2, entry 13). Heating the reaction mixture to 50 °C enabled the reaction to be completed in a short time (2 h) but gave similar yield of the desired product (Table 3, entry 1). The absence of base and additive and ability to perform the oxidation at ambient temperature makes the protocol a greener procedure.
|
|||
|---|---|---|---|
| Entry | Deviation from “standard conditions” | Time (h) | Yieldb (%) |
| a Reaction condition: 4-methoxybenzyl alcohol 6a (0.5 mmol), CuBr (5 mol%), FL2 (5 mol%), TEMPO (5 mol%), ambient air, water (0.5 mL), r.t.b Isolated yields. Purity is >95% (according to NMR).c Recovered starting material. | |||
| 1 | None | 12 | 90 |
| 2 | FeCl2·H2O | 12 | 6 |
| 3 | FeCl3 | 12 | 26 |
| 4 | FeSO4·H2O | 12 | Trace |
| 5 | Fe(C2O4)·H2O | 12 | Trace |
| 6 | Absence of TEMPO | 12 | — |
| 7 | Under inert atm | 12 | Trace |
| 8 | Absence of FL2 | 12 | 57 (13c) |
| 9 | 10 mol% FL2 | 6 | 29 |
| 10 | 3 mol% CuBr, 3 mol% FL2 and 3 mol% TEMPO | 7 | 43 |
| 11 | 1 mol% CuBr, 1 mol% FL2 and 1 mol% TEMPO | 7 | 40 |
| 12 | 3 mol% CuBr and 3 mol% FL2 | 5 | 53 |
| 13 | None | 5 | 91 |

|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Time (h) | Yieldb (%) |
| a Reaction condition: substrate (0.5 mmol), CuBr (5 mol%), FL2 (5 mol%), TEMPO (5 mol%), ambient air, water (0.5 mL), r.t.b Isolated yields. Purity is >95% (according to NMR).c Oil bath temperature of 50 °C.d Using CuCl/DMAP:9k 5 mol% CuCl, 10 mol% DMAP, 5 mol% TEMPO, H2O, air, r.t.e Using pytl-β-CD:9h 5 mol% Cu(OAc)2·H2O, 5 mol% pytl-β-CD, 5 mol% TEMPO, Na2CO3, H2O, air, reflux.f Using CuCl/DMAP:9k 5 mol% CuCl, 10 mol% DMAP, 5 mol% TEMPO, H2O, air, 55 °C.g Recovered starting material. | ||||
| 1 |  |
 |
5 (2c, 10d,e) | 91 (93c,d,e) |
| 2 |  |
 |
5 (5d, 10e) | 90 (95d, 94e) |
| 3 |  |
 |
6 | 84 |
| 4 |  |
 |
24 (10d) | 73 (90d) |
| 5 |  |
 |
7 (10e, 10f) | 86 (93e, 95f) |
| 6 |  |
 |
12 (12e) | 78 (95e) |
| 7 | 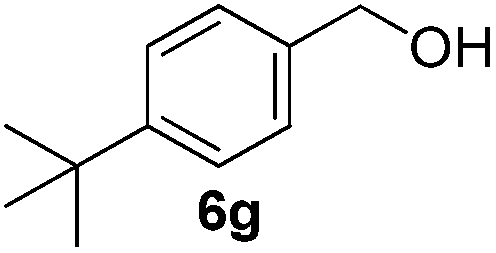 |
 |
5 | 85 |
| 8 | 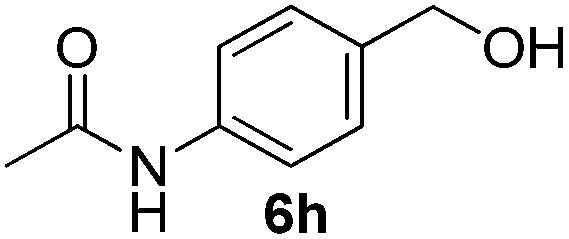 |
 |
24 | 71 |
| 9 | 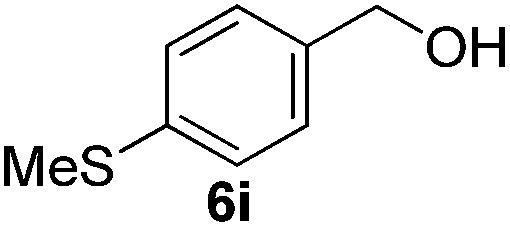 |
 |
24 | 76 |
| 10 | 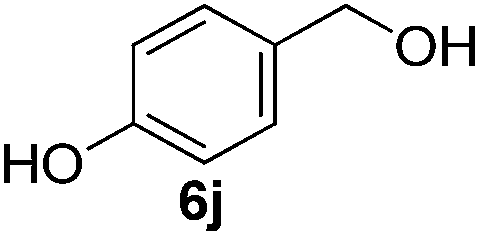 |
 |
7 | 67 |
| 11 |  |
 |
10 | 59 |
| 12 | 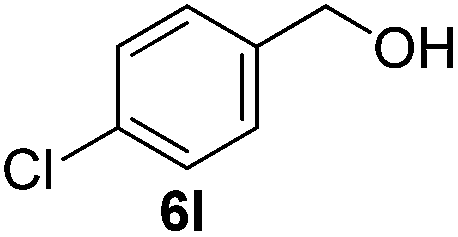 |
 |
8 (10d, 10e) | 96 (94d, 93e) |
| 13 |  |
 |
8 (16e) | 61 (92e) |
| 14 |  |
 |
8 (14e) | 60 (93e) |
| 15 |  |
 |
8 | 61 |
| 16 | 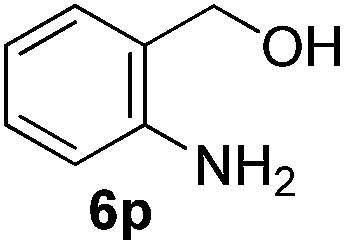 |
 |
8 | 73 |
| 17 |  |
 |
10 (12e, 10f) | 79 (80e, 92f) |
| 18 |  |
 |
12 | 63 (10g) |
| 19 |  |
 |
10 | 76 |
| 20 |  |
 |
13 (10e) | 88 (58e) |
| 21 |  |
 |
9 | 91 |
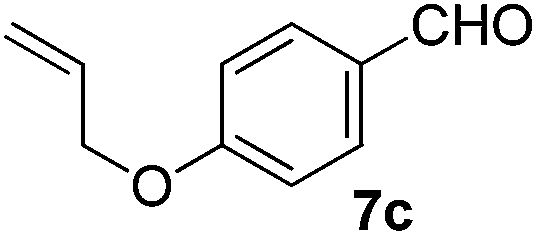
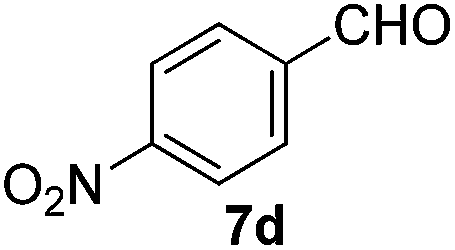
Having established the optimal reaction conditions, several benzylic alcohols were tested to explore the generality of the transformation (Table 3). The reaction conditions were compatible with both electron-withdrawing and -donating substituents (Table 3, entries 1–9, 12). Substrate 6c bearing reactive and coordinating alkene functional group did not affect the reaction and the corresponding aldehyde 7c was obtained in 84% yield (Table 3, entry 3). The reaction condition was also amenable to substrates with acidic protons albeit slightly lower yields were obtained (Table 3, entries 10 and 11). The size and electronic effect of halogen substituent (I, Br, Cl) did not affect the transformation and provided the respective products in similar yields (Table 3, entries 13–15). However, the transformation is sensitive to steric effects which could be observed from the higher yield obtained with 4-chlorobenzyl alcohol (96%, Table 3, entry 12) as compared to 2-chlorobenzyl alcohol (61%, Table 3, entry 13). Heteroaromatic substrates were also compatible (Table 3, entries 17–19) with the aqueous procedure although substrate 6q (Table 3, entry 17) provided the product in lower yield than the pytl-β-CD/Cu(OAc)2 (ref. 9h) system which nevertheless required the presence of a base in refluxing conditions. The oxidation of unprotected indole 6r was unable to reach completion even after prolonged reaction time (Table 3, entry 18). Besides benzylic alcohols, allylic alcohols were also successfully oxidized to the respective aldehydes in excellent yields (Table 3, entries 20 and 21).
Besides primary benzylic and allylic alcohols, we have also applied the reaction to a variety of secondary benzylic alcohols (Table 4). These latter alcohols are less reactive and required mild heating (50 °C) to provide the desired ketone in good to moderate yields. The less reactive secondary benzylic alcohol 8a provided compound 9a in 88% yield (Table 4, entry 1) which is slightly lower than that reported using CuCl/DMAP and 9-azabicyclo[3.3.1]nonane N-oxyl (ABNO),9k a relatively more expensive nitroxyl radical than TEMPO.
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Time (h) | Yieldb (%) |
| a Reaction condition: substrate (0.5 mmol), CuBr (5 mol%), FL2 (5 mol%), TEMPO (5 mol%), ambient air, water (0.5 mL), 50 °C.b Isolated yields. Purity is >95% (according to NMR).c Using CuCl/DMAP:9k 5 mol% CuCl, 10 mol% DMAP, 5 mol% ABNO, H2O, air, r.t. | ||||
| 1 |  |
 |
12 (5c) | 88 (96c) |
| 2 |  |
 |
24 | 81 |
| 3 |  |
 |
24 | 76 |
| 4 |  |
 |
12 | 90 |

The difference in reaction rates between primary and secondary benzylic alcohols allows for the selectively oxidation of primary benzylic alcohols. Thus to investigate the selective oxidation of primary benzylic alcohol, compound 6v was subjected to the optimized reaction condition. Compound 7v was isolated in 92% yield with trace amount (5%) of the doubly oxidized product 7v′ (Scheme 2). The selectivity experiment was extended to compound 6w which carries an aliphatic alcohol substituent. Similarly, compound 7w was obtained as the major product (86%) with the dialdehyde 7w′ present in only 8% yield. To demonstrate the usefulness of the optimized condition, Gram scale oxidation of 4-methoxybenzyl alcohol 6a was carried out and the desired product 4-methoxybenzaldehyde 7a was obtained solely after 6 h (complete conversion indicated by TLC) in 89% yield. The ability of recovering FL2 in good yield (94%) adds to the effectiveness of the procedure for Gram-scale oxidation.
 | ||
| Scheme 2 Selectivity experiments and Gram-scale synthesis of 7a. | ||
Copper-catalyzed allylic and benzylic sp3 C–H oxidation
We have previously reported6c a copper-catalyzed allylic and benzylic sp3 C–H oxidation in water using T-Hydro as oxidant and SDS as surfactant. To determine if our fluorous bispidine-type ligand could be applied to such oxidations, we carried out the initial assessment by employing the reaction condition from our previous studies.6c Using 1-phenyl-1-cyclohexene 10a as the model substrate, with 5 mol% CuI as the metal catalyst, 5 mol% L3 as ligand, 5 mol% SDS as surfactant, 5 equivalent of T-Hydro as oxidant and water as solvent at room temperature, the desired product 11a was obtained in 45% yield after 12 h (Table 5, entry 1). Varying the oxidant and introducing a base (KOAc) lowered the yield whilst the absence of SDS increased the yield to 53% (Table 5, entries 2–5). The absence of SDS compared to our previously reported condition increases the greenness of the current procedure. Next, we varied the metal catalyst but CuI was most efficient for this transformation (Table 5, entries 6–13). Iron salts were also employed as catalyst but other than Fe(OTf)2, the reactions were unable to reach completion (Table 5, entries 10–13). Subsequently, we varied the ligands and found that L1 and L3 provided the product in similar yields while L2 gave a slightly lower yield (Table 5, entries 5, 14–15). Since L1 is structurally similar to the previously employed FL2, we decided to use it for the remaining optimization process.
|
|||
|---|---|---|---|
| Entry | Metal catalyst | Ligand | Yieldb (%) |
| a Reaction condition: 1-phenyl-1-cyclohexene (0.5 mmol), Cu salt (5 mol%), ligand (5 mol%), T-Hydro (5 equiv.), water (1.0 mL), r.t., 12 h.b Isolated yields. Purity is >95% (according to NMR).c 5 mol% SDS added.d 5 mol% SDS added and using H2O2 instead of T-Hydro.e 5 mol% SDS and 0.5 equiv. KOAc added.f 0.5 equiv. KOAc added.g Recovered starting material. | |||
| 1c | CuI | L3 | 45 |
| 2d | CuI | L3 | — |
| 3e | CuI | L3 | 42 |
| 4f | CuI | L3 | 35 |
| 5 | CuI | L3 | 53 |
| 6 | CuBr | L3 | 41 |
| 7 | CuBr2 | L3 | 45 |
| 8 | Cu(OAc)2 | L3 | 36 |
| 9 | Cu(OTf)2 | L3 | 17 |
| 10 | FeCl2·4H2O | L3 | 28 (52g) |
| 11 | FeCl3 | L3 | 26 (49g) |
| 12 | Fe(ClO4)2·6H2O | L3 | 27 (54g) |
| 13 | Fe(OTf)2 | L3 | 40 |
| 14 | CuI | L2 | 49 |
| 15 | CuI | L1 | 53 |

Next, we varied the amount of T-Hydro used and determined that the reaction is most efficient with 5 equivalents of T-hydro (Table 6, entries 2–4). The amount of CuI and L1 used were also investigated and 5 mol% of CuI and 5 mol% of L1 were found to be the optimal amounts required (Table 6, entries 5 and 6). To recover and recycle the ligand, we replaced the non-fluorous L1 with the three fluorous ligands developed. Coincidently, FL1 was most efficient in the aqueous procedure giving compound 11a in 69% yield (Table 6, entries 7–9). FL2, which was previously used in the benzylic alcohol oxidation, was also compatible with the allylic oxidation reaction giving comparable yields to FL1 (Table 6, entry 8). A control experiment conducted in the absence of FL1 yielded only trace amount of the desired product (Table 6, entry 10).
|
||
|---|---|---|
| Entry | Deviation from “standard conditions” | Yieldb (%) |
| a Reaction condition: 1-phenyl-1-cyclohexene (0.5 mmol), CuI (5 mol%), L1 (5 mol%), T-Hydro (5 equiv.), water (1.0 mL), r.t., 12 h.b Isolated yields. Purity is >95% (according to NMR). | ||
| 1 | None | 53 |
| 2 | 6 equivalents T-Hydro | 40 |
| 3 | 4 equivalents T-Hydro | 32 |
| 4 | 3 equivalents T-Hydro | 32 |
| 5 | 1 mol% CuI and 1 mol% L1 | 7 |
| 6 | 10 mol% CuI and 10 mol% L1 | 42 |
| 7 | 5 mol% FL3 instead of L1 | 58 |
| 8 | 5 mol% FL2 instead of L1 | 63 |
| 9 | 5 mol% FL1 instead of L1 | 69 |
| 10 | Absence of FL1 | Trace |

Next, we investigated the generality of the optimized conditions for allylic and benzylic oxidation with various alkenes and alkylarenes (Table 7). Moderate to good yields were obtained for the allylic oxidation of cycloalkenes containing both electron-withdrawing and donating-groups (Table 7, entries 1–5). However the yields were generally lower than the yields obtained by our previously reported6c procedure (Table 7, entries 1–3). Next, we examined the oxidation reaction of benzylic substrates (Table 7, entries 6–13). Ethylbenzene was successfully oxidized to acetophenone in 80% yield which was higher than the yield obtained via our previously reported procedure (Table 7, entry 6). Alkylarene with a longer alkyl chain 10g resulted in a lower yield (Table 7, entry 7) but cyclic alkylarene 10h was successfully oxidized under the aqueous procedure to give compound 11g in good yield (Table 7, entry 8). The oxidation of fluorene 10i at room temperature was sluggish and required a higher temperature (50 °C). The requirement for higher temperature was also observed for solid substrates (Table 7, entries 9 and 12). The presence of electron-withdrawing methyl ester functionality on the alkyl moiety of the alkylarene causes the benzylic C–H to be inert and thus resulting in substrate 10j being a challenging substrate for oxidation.20 Gratifyingly, oxidation of substrate 10j was successfully carried out under our optimized conditions giving product 11i in 80% yield without any hydrolyzed product (Table 7, entry 10). Heteroatom bearing substrates were also compatible with the reaction condition and provided the corresponding oxidized product in good yields (Table 7, entries 11 and 12). Oxidation of amine 10m yielded amide 11l in 61% yield (Table 7, entry 13).
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yieldb (%) |
| a Reaction condition: substrate (0.5 mmol), CuI (5 mol%), FL1 (5 mol%), T-Hydro (5 equiv.), water (1.0 mL), r.t., 12 h.b Isolated yield. Purity is >95% (according to NMR).c Using CuI/fluorous tridentate ligand:6c 5 mol% CuI, 5 mol% fluorous tridentate ligand, 5 mol% SDS, T-Hydro, water, r.t., 1 h.d Oil bath temperature of 50 °C.e Using CuI/fluorous tridentate ligand:6c 5 mol% CuI, 5 mol% fluorous tridentate ligand, 5 mol% SDS, T-Hydro, water, 50 °C, 2 h.f Using CuI/fluorous tridentate ligand:6c 5 mol% CuI, 5 mol% fluorous tridentate ligand, 5 mol% SDS, T-Hydro, water, 50 °C, 3 h. | |||
| 1 |  |
 |
69 (71c) |
| 2 |  |
 |
36 (62c) |
| 3 |  |
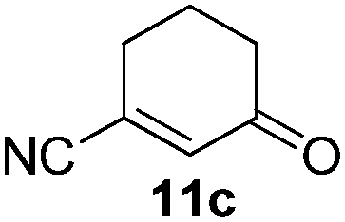 |
46 (74c) |
| 4 |  |
 |
45 |
| 5 |  |
 |
39 |
| 6 |  |
 |
80 (68c) |
| 7 |  |
 |
41 (51c) |
| 8 |  |
 |
74 (70c) |
| 9d |  |
 |
93 (90e) |
| 10 |  |
 |
80 (73c) |
| 11 | 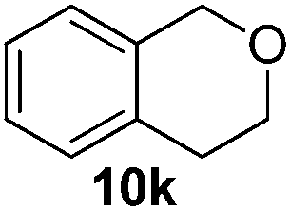 |
 |
75 (68c) |
| 12d |  |
 |
73 (51f) |
| 13 | 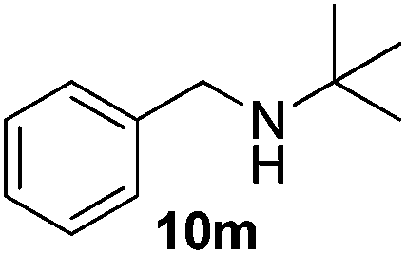 |
 |
61 |

Next, to determine the applicability of the reaction, we carried out a Gram-scale oxidation using our optimized conditions and 1.58 g (10 mmol) of 10a (Scheme 3). The reaction was completed in 12 h (indicated by TLC) and 1.18 g (69%, 6.86 mmol) of 11a was obtained with 10% of 11a′ as side product and 97% of FL1 was recovered via F-SPE. Previously, we proposed a radical mechanism for the copper-catalyzed allylic oxidation using T-Hydro.6c The presence of the tert-butylperoxy intermediate 11a′ and the subsequent oxidation of 11a′ to give product 11a under the optimized condition supported the proposed radical mechanism. Furthermore, the addition of 2,6-di-tert-butyl-4-methylphenol (BHT) to the reaction system ceased the reaction, lending support to the radical mechanism.
 | ||
| Scheme 3 Gram-scale synthesis of 11a and mechanistic experiments. | ||
Knoevenagel condensation
The Knoevenagel condensation reaction is a classical method used for the formation of C–C bond in the synthesis of benzylidene compounds. The condensation of benzylic aldehydes with malononitrile furnishes benzylidene malononitrile derivatives which are important building blocks in cyclization reactions,21 tumour cytotoxic agents and rodent and riot control agents.22 Herein we examined the applicability of our fluorous bispidine-type ligand as a proton sponge to the base-catalyzed Knoevenagel condensation reaction. The proof of concept was conducted using benzaldehyde 7b and malononitrile with 2 mol% of L3 as a base and water as solvent at 40 °C. The desired benzylidene 13a was obtained after 2 h in 75% yield (Table 8, entry 1). We sought to use the same ligand for both aerobic oxidation and Knoevenagel condensation to demonstrate the dual purpose of the ligand. Thus, we conducted the condensation reaction under the same condition using L2 and a similar yield (78%) was obtained (Table 8, entry 2). Encouraged by these results, we proceeded to replace L2 with FL2 and gratifyingly, 97% of the benzylidene 13a was obtained (Table 8, entry 3). Purification of the product was also simplified with the use of fluorous base FL2 as no extraction or column chromatography was required. The desired product and FL2 could be easily separated via F-SPE (See Experimental section for details). Decreasing the reaction temperature (room temperature) lengthened the reaction time with a slight decrease in yield (Table 8, entry 4). Lowering the amount of FL2 to 1 mol% did not result in any observable change but further reduction to 0.5 mol% provided in a slight decrease in yield (Table 8, entries 5 and 6).
|
||||
|---|---|---|---|---|
| Entry | Proton sponge (mol%) | Temp (°C) | Time (h) | Yieldb (%) |
| a Reaction condition: benzaldehyde 7b (0.5 mmol), malononitrile (0.5 mmol), proton sponge, water (5.0 mL).b Isolated yield. Purity is >95% (according to NMR).c Isolated using column chromatography. | ||||
| 1c | L3 (2 mol%) | 40 | 2 | 75 |
| 2c | L2 (2 mol%) | 40 | 2 | 78 |
| 3 | FL2 (2 mol%) | 40 | 2 | 97 |
| 4 | FL2 (2 mol%) | r.t. | 6 | 88 |
| 5 | FL2 (1 mol%) | 40 | 2 | 96 |
| 6 | FL2 (0.5 mol%) | 40 | 2 | 91 |

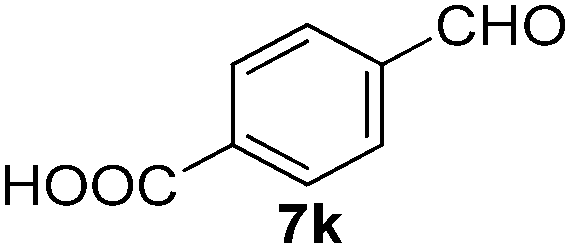
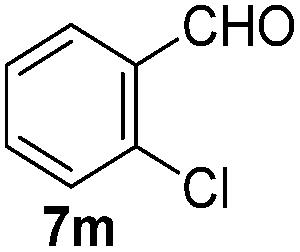
With the optimized reaction conditions, the generality of the reaction was explored (Table 9). The reaction condition was amenable to substrates containing both electron-withdrawing and -donating substituents as well as those bearing acidic protons (Table 9, entries 1–9). A comparison with other Knoevenagel condensation reactions carried out in water showed that the yields obtained with FL2 were comparable to those using supported bases (Table 9, entry 1). 4-Nitrobenzaldehyde (Table 9, entry 3) provided a lower yield of the desired product than the other aldehydes bearing heteroaromatic or electron-donating groups. This trend could be attributed to the effect of the electron-withdrawing group at the para-position. When the electron-withdrawing cyano group is in the meta-position, compound 13i was obtained in excellent yield (Table 9, entry 9), implying that the reaction is affected by the position of the electron-withdrawing group. However steric factor does not appear to affect the condensation reaction since both 2-chlorobenzaldehyde 7m and 4-chlorobenzaldehyde 7l provided the respective product in similar yields (Table 9, entries 4 and 8). Allylic, heteroaromatic and aliphatic aldehydes are also compatible with the optimized condition, demonstrating the applicability of the transformation to different classes of aldehyde (Table 9, entries 10–14). Encouraged by the results obtained, we extended the reaction to the less reactive ketones. However under the optimized condition, the product was obtained in moderate yield (Table 9, entries 15–16).
|
||||
|---|---|---|---|---|
| Entry | Aldehyde/ketone | Product | Time (h) | Yieldb (%) |
| a Reaction condition: aldehyde/ketone (0.5 mmol), malononitrile (0.5 mmol), FL2 (1 mol%), water (5.0 mL), 40 °C.b Isolated yield. Purity is >95% (according to NMR).c Using C/Co@DMAN:14b 2 mol% C/Co@DMAN, H2O, r.t., 7.5 h.d Using SBILs:14c 25 mg SBILs, H2O, 30 °C, 1 h. | ||||
| 1 | 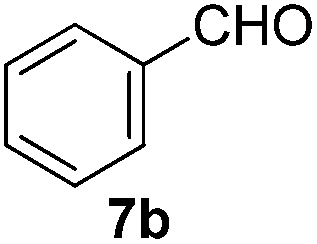 |
 |
2 | 96 (97c, 90d) |
| 2 |  |
 |
2 | 95 |
| 3 |  |
 |
2 | 71 |
| 4 |  |
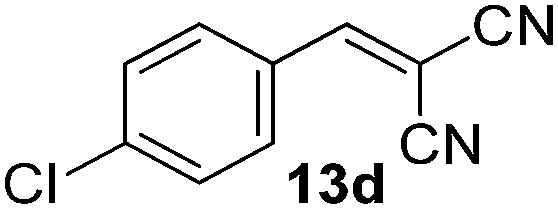 |
2 | 96 |
| 5 |  |
 |
2 | 95 |
| 6 | 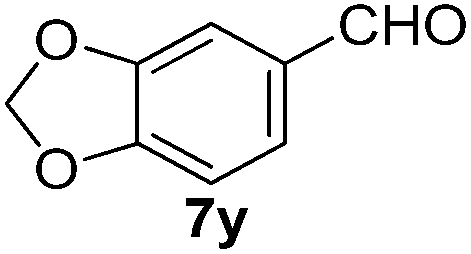 |
 |
2 | 92 |
| 7 |  |
 |
2 | 98 |
| 8 |  |
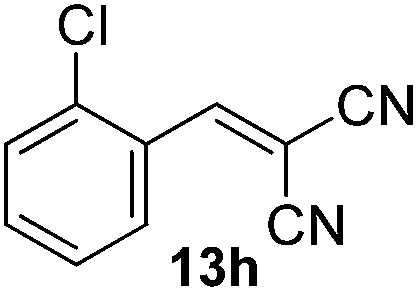 |
2 | 94 |
| 9 | 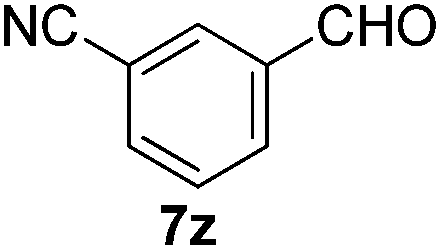 |
 |
2 | 91 |
| 10 |  |
 |
4 | 83 |
| 11 |  |
 |
2 | 93 |
| 12 |  |
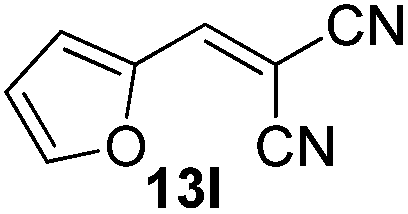 |
2 | 90 |
| 13 |  |
 |
2 | 79 |
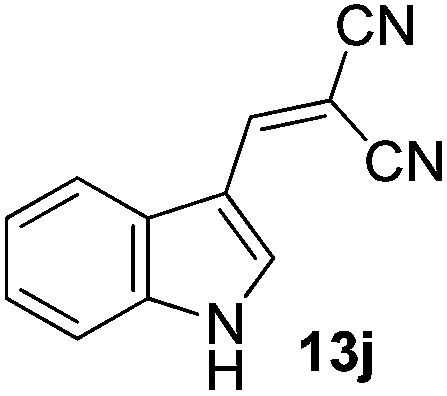
| 14 |  |
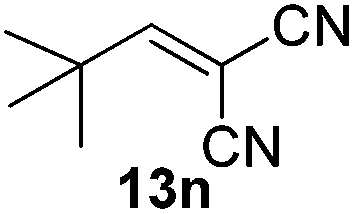 |
2 | 88 |
| 15 |  |
 |
6 | 46 |
| 16 |  |
 |
2 | 52 |

Besides malononitrile, we investigated the use of other activated methylenes in this transformation (Table 10). We varied the activated methylene by replacing one of the cyano groups in malononitrile with either an ester, amide or ketone and the experimental data obtained indicated that good to excellent yields of the desired product was achieved with the optimized condition (Table 10, entries 1–3). Nitro-substituted phenyl group was also employed albeit the reaction needed higher reaction temperature (60 °C) and longer reaction time (24 h). The desired product 13t was obtained in moderate yields (Table 10, entry 4).




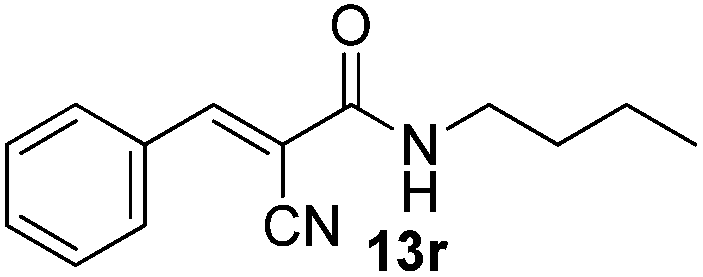




Compound 13u is used in sunscreen compositions containing a UV-A sunscreen, photostabilizer and antioxidant.23 The compound was initially synthesized under refluxing condition with acetic acid/benzene as solvent and piperidine as catalyst which gave the product in 95% yield.23a Later, Gomes et al.23b improved the green aspect of the reaction by performing the reaction at room temperature, replacing the solvent with water and using morpholine as the catalyst. This afforded 13u in 85% yield. The synthesis of 13u could also be achieved with our aqueous protocol. By treating vanillin 7ac with ethyl cyanoacetate 12b under the optimized condition, 13u was obtained in 96% yield (Scheme 4).
 | ||
| Scheme 4 Synthesis of compound 13u. | ||
Tandem oxidation/Knoevenagel condensation
Since both the aerobic oxidation and the Knoevenagel condensation were carried out in water and utilized the same ligand/base FL2, we envisioned the possibility of conducting a tandem oxidation/Knoevenagel condensation reaction. Benzyl alcohol 6b was subjected to aerobic oxidation under the optimized conditions. Upon completion of the oxidation reaction (as determined by TLC), malononitrile was added and the reaction mixture was further reacted under the optimized Knoevenagel reaction condition to yield benzylidene 13a in 79% yield (over 2 steps, Table 11, entry 1). As anticipated, the reaction was successfully conducted with benzyl alcohols containing electron-withdrawing and donating-groups (Table 11, entries 2–7) as well as allylic and heteroaromatic substrates (Table 11, entries 8–10). The less reactive secondary benzylic alcohol 1-phenylethanol 8a was also subjected to the tandem procedure and provided the product 13o in 42% yield (over 2 steps, Table 11, entry 11). The yields obtained from the tandem reaction were generally comparable or slightly lower than the combined yields obtained when both reactions were carried out and purified independently (Table 11, entries 1, 3–4, 7–8 and 11). Notwithstanding, a tandem reaction eliminates intermittent work-up and purification thus saving time, energy and the excessive usage of solvents, which ultimately leads to a greener procedure.
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Time, t1 (h) | Yieldb (%) |
| a Reaction condition: 1. Benzylic alcohol (0.5 mmol), CuBr (5 mol%), FL2 (5 mol%), TEMPO (5 mol%), ambient air, H2O (0.5 mL), r.t.; 2. Malononitrile (0.5 mmol), H2O (4.5 mL), 40 °C, 2 h.b Isolated yields over 2 steps. Purity is >95% (according to NMR).c Combined isolated yields if both steps were carried out separately.d Using Pd1–Au1/LDH:15a 1. 1.5 mol% Pd1–Au1/LDH, H2O, O2 balloon, 80 °C, 1.5 h; 2. 80 °C, 1 h.e 6 h required for second step. | ||||
| 1 |  |
 |
5 | 79 (86c, 94.6d) |
| 2 |  |
 |
7 | 73 |
| 3 |  |
 |
5 | 80 (86c) |
| 4 |  |
 |
8 | 85 (92c) |
| 5 |  |
 |
5 | 75 |
| 6 |  |
 |
12 | 68 |
| 7 |  |
 |
24 | 49 (52c) |
| 8 |  |
 |
13 | 66 (70c) |
| 9 |  |
 |
10 | 74 |
| 10 |  |
 |
10 | 67 |
| 11 | 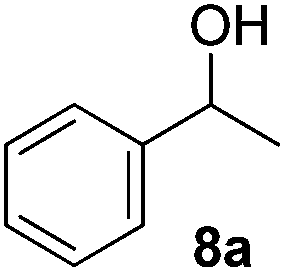 |
 |
12 + 6e | 42 (40c) |

Recycling of fluorous ligands
Finally, we investigated the possibility of recovering and reusing FL2 (Table 12). 4-Methoxybenzyl alcohol 6a was used as the model substrate under the optimized condition for the copper-catalyzed aerobic oxidation reaction. We found that over 5 runs, the time taken for the reaction to complete was 5–7 h and aldehyde 7a was obtained in 85–90% yields with 92–96% of FL2 recovered (Table 12, entries 1–5). To investigate the possibility of recycling and reusing FL1, substrate 10a was used as the model substrate under the optimized reaction condition. The recycling experiments were carried out over five cycles and the product 11a was obtained in 60–69% yield with 92–97% of FL1 recovered via F-SPE (Table 12, entries 6–10). A slow decline of the catalytic activity of FL1 was observed after several cycles. Similarly, to address the recyclability of FL2 in the Knoevenagel condensation reaction, the recycling experiments were carried out with benzaldehyde 7b and malononitrile as model substrates. The results obtained were gratifying as the yields obtained over five runs were quantitative and the time required for the reaction to go to completion remained short. In addition, the recovery of FL2 was 92–97% (Table 12, entries 11–15). Finally, recycling experiments were carried out to determine the recyclability of FL2 in the tandem oxidation/Knoevenagel condensation reaction. The recycling experiments were carried out over five cycles and the product 13a was obtained in 73–79% yield (over 2 steps) with 91–96% of FL2 recovered (Table 12, entries 16–20).| Entry | Cycle | Time (h) | Yieldb (%) | Recovered FL1/FL2c (wt%) |
|---|---|---|---|---|
| a Reaction condition: respective optimized reaction conditions (2.5 mmol scale for aerobic oxidation, allylic oxidation and tandem reaction; 16 mmol scale for Knoevenagel condensation).b Isolated yields. Purity is >95% (according to NMR).c Recovered via F-SPE.d Refers to time t1 + t2.e Isolated yields over 2 steps. Purity is >95% (according to NMR). | ||||
 |
||||
| 1 | 1 | 5 | 90 | 96 |
| 2 | 2 | 5 | 89 | 93 |
| 3 | 3 | 6 | 86 | 94 |
| 4 | 4 | 6 | 87 | 94 |
| 5 | 5 | 7 | 85 | 92 |
 |
||||
| 6 | 1 | 12 | 69 | 97 |
| 7 | 2 | 12 | 68 | 96 |
| 8 | 3 | 12 | 65 | 94 |
| 9 | 4 | 12 | 61 | 92 |
| 10 | 5 | 12 | 60 | 93 |
 |
||||
| 11 | 1 | 2 | 96 | 97 |
| 12 | 2 | 2 | 95 | 97 |
| 13 | 3 | 3 | 94 | 95 |
| 14 | 4 | 3 | 95 | 93 |
| 15 | 5 | 3 | 93 | 92 |
 |
||||
| 16 | 1 | 5 + 2d | 79e | 96 |
| 17 | 2 | 5 + 2d | 77e | 97 |
| 18 | 3 | 6 + 3d | 76e | 94 |
| 19 | 4 | 7 + 3d | 74e | 93 |
| 20 | 5 | 7 + 3d | 73e | 91 |
Experimental
General
All chemicals purchased were used without further purification. Compound 1,16 3,18 4,19 L217 and p26c were synthesized according to previously reported procedures. Moisture-sensitive reactions were carried out under nitrogen with commercially obtained anhydrous solvents. Analytical thin-layer chromatography (TLC) was carried out on precoated F254 silica plates and visualized with UV light. Column chromatography was performed with silica (Merck, 230–400 mesh). F-SPE was performed with FluoroFlash® silica gel (40 micron). 1H and 13C NMR spectra were recorded at 298 K. Chemical shifts are expressed in terms of δ (ppm) relative to the internal standard tetramethylsilane (TMS). Mass spectra were performed under EI and ESI mode.Synthesis of ligands
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (5.0 mL) was added NaBH4 (95 mg, 2.5 mmol). Addition portion of NaBH4 was added every 2 h (total 3 portions) and the reaction mixture was stirred at room temperature for 12 h. The reaction mixture was quenched at 0 °C with 50% aqueous HCl and allowed to stir at room temperature for 30 min. 20% NaOH was added to the reaction mixture to pH 10 and consecutively washed with DCM (20 mL × 3). The combined organic layer was washed with water (10 mL × 3) and brine (10 mL × 3), dried over anhydrous MgSO4, filtered and concentrated. The desired product 5 (223 mg, 94%) was obtained as a white solid and used without further purification. To a solution of compound 5 (223 mg, 0.47 mmol) in THF (1.0 mL) was added NaH (60% in mineral oil, 38 mg, 0.94 mmol) and R–I (0.94 mmol). The reaction mixture was refluxed for 12 h, quenched with MeOH (5 mL) and purified by column chromatography.
:1 (5.0 mL) was added NaBH4 (95 mg, 2.5 mmol). Addition portion of NaBH4 was added every 2 h (total 3 portions) and the reaction mixture was stirred at room temperature for 12 h. The reaction mixture was quenched at 0 °C with 50% aqueous HCl and allowed to stir at room temperature for 30 min. 20% NaOH was added to the reaction mixture to pH 10 and consecutively washed with DCM (20 mL × 3). The combined organic layer was washed with water (10 mL × 3) and brine (10 mL × 3), dried over anhydrous MgSO4, filtered and concentrated. The desired product 5 (223 mg, 94%) was obtained as a white solid and used without further purification. To a solution of compound 5 (223 mg, 0.47 mmol) in THF (1.0 mL) was added NaH (60% in mineral oil, 38 mg, 0.94 mmol) and R–I (0.94 mmol). The reaction mixture was refluxed for 12 h, quenched with MeOH (5 mL) and purified by column chromatography.General procedure for the copper-catalyzed aerobic oxidation of alcohols
A mixture of the substrate (0.5 mmol), FL2 (5 mol%), CuBr (5 mol%), TEMPO (5 mol%) and water (0.5 mL) was stirred at room temperature (50 °C for secondary alcohols) in the presence of ambient air. The reaction was monitored by TLC. When the reaction has completed, the reaction mixture was diluted with EtOAc (20 mL) and the organic layer was separated. The aqueous layer was further extracted with EtOAc (20 mL). The combined organic layer was dried over anhydrous MgSO4, filtered and concentrated. The crude product was then purified by passing through a short pad of silica.General procedure for the copper-catalyzed allylic and benzylic oxidation
A mixture of the substrate (0.5 mmol), FL1 (5 mol%), CuI (5 mol%) and water (1.0 mL) was stirred at room temperature. T-Hydro (5 equiv.) was added dropwise to the stirred solution and the reaction mixture was allowed to further stirred at room temperature for 12 h. The reaction was monitored by TLC. When the reaction has completed, the reaction mixture was diluted with EtOAc (20 mL) and the organic layer was separated. The aqueous layer was further extracted with EtOAc (20 mL). The combined organic layer was dried over anhydrous MgSO4, filtered and concentrated. The crude product was then purified by column chromatography.General procedure for the base-catalyzed Knoevenagel condensation
A mixture of the respective aldehyde (0.5 mmol), malononitrile (0.5 mmol), FL2 (1 mol%) and water (5.0 mL) was stirred at 40 °C. The reaction was monitored by TLC. When the reaction has completed, water was removed and the crude product was purified by F-SPE. The crude product was first diluted with THF–H2O = 7:3 (1 mL) and loaded into an F-SPE fluorous silica column (column diameter: 2.20 cm; amount of fluorous silica required: 10% of crude product by weight). The pure product was then eluted using THF–H2O = 7:3 (30 mL) as eluent.
General procedure for the tandem oxidation/Knoevenagel condensation
A mixture of the substrate (0.5 mmol), FL2 (5 mol%), CuBr (5 mol%), TEMPO (5 mol%) and water (0.5 mL) was stirred at room temperature in the presence of ambient air. The reaction was monitored by TLC. When the reaction has completed, malononitrile (0.5 mmol) and water (4.5 mL) was added to the reaction mixture and stirred at 40 °C. The reaction was monitored by TLC. When the reaction has completed, the reaction mixture was diluted with EtOAc (20 mL) and the organic layer was separated. The aqueous layer was further extracted with EtOAc (20 mL). The combined organic layer was dried over anhydrous MgSO4, filtered and concentrated. The crude product was then purified by column chromatography.General procedure for the recycling experiment of aerobic oxidation using F-SPE
Recycling experiments were carried out on a 2.50 mmol scale using the general procedure described above. The crude product was first diluted with THF–H2O = 7:3 (1 mL) and loaded into a F-SPE fluorous silica column (column diameter: 2.20 cm; amount of fluorous silica required: 10% of crude product by weight). The crude product was then eluted using THF–H2O = 7:3 (30 mL) as eluent and FL2 was subsequently eluted with THF (20 mL). The fluorous silica was regenerated and reused (5 times) after washing with acetone (10 mL). To determine the amount of product formed after each recycling experiment, the solution of pure product was concentrated and then purified accordingly.
General procedure for the recycling experiment of the copper-catalyzed allylic and benzylic oxidation using F-SPE
Recycling experiments were carried out on a 2.50 mmol scale using the general procedure described above. The crude product was first diluted with THF–H2O = 7:3 (1 mL) and loaded into a F-SPE fluorous silica column (column diameter: 2.20 cm; amount of fluorous silica required: 10% of crude product by weight). The crude product was then eluted using THF–H2O = 7:3 (30 mL) as eluent and FL1 was subsequently eluted with THF (20 mL). The fluorous silica was regenerated and reused (5 times) after washing with acetone (10 mL). To determine the amount of product formed after each recycling experiment, the solution of crude product was concentrated, diluted with EtOAc (20 mL) and washed with water (10 mL). The organic layer was dried over anhydrous MgSO4, filtered, concentrated and then purified accordingly.
General procedure for the recycling experiment of Knoevenagel condensation using F-SPE
Recycling experiments were carried out on a 16.0 mmol scale using the general procedure described above. The crude product was first diluted with THF–H2O = 7:3 (5 mL) and loaded into a F-SPE fluorous silica column (column diameter: 2.20 cm; amount of fluorous silica required: 10% of crude product by weight). The pure product was then eluted using THF–H2O = 7:3 (150 mL) as eluent and FL2 was subsequently eluted with THF (100 mL). The fluorous silica was regenerated and reused (5 times) after washing with acetone (50 mL). To determine the amount of product formed after each recycling experiment, the solution of pure product was concentrated.
General procedure for the recycling experiment of tandem oxidation/Knoevenagel condensation using F-SPE
Recycling experiments were carried out on a 2.50 mmol scale using the general procedure described above. The crude product was first diluted with THF–H2O = 7:3 (1 mL) and loaded into a F-SPE fluorous silica column (column diameter: 2.20 cm; amount of fluorous silica required: 10% of crude product by weight). The crude product was then eluted using THF–H2O = 7:3 (30 mL) as eluent and FL2 was subsequently eluted with THF (20 mL). The fluorous silica was regenerated and reused (5 times) after washing with acetone (10 mL). To determine the amount of product formed after each recycling experiment, the solution of pure product was concentrated and then purified accordingly.
Conclusions
In summary, fluorous bispidine-type ligands FL1 to FL3 and non-fluorous bispidine-type ligands L1 to L3 were synthesized and evaluated for their activities as ligands for the copper-catalyzed aerobic oxidation of benzylic and allylic alcohols and the copper-catalyzed allylic and benzylic sp3 C–H oxidation reaction. FL2 was shown to, not only, promote oxidation reactions in water but was capable of serving as a proton sponge in the Knoevenagel condensation reaction. The dual function of FL2 was further demonstrated in the tandem oxidation/Knoevenagel condensation in water and allowed the tandem reaction to be carried out without intermittent workup and purification. Recycling was also possible with FL2 which was reused five times without significant loss of activity. FL1 was also amenable to the sp3 C–H oxidation reaction in water and could be reused five times without significant loss of activity.Acknowledgements
This research is supported by a grant from the Ministry of Education (Singapore) (MoE Tier 2: R-143-000-589-112).Notes and references
-
(a) M. Sono, M. P. Roach, E. D. Coulter and J. J. Dawson, Chem. Rev., 1996, 96, 2841 CrossRef CAS PubMed
; (b) M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Chem. Rev., 2004, 104, 939 CrossRef CAS PubMed
-
(a) J. Bautz, P. Comba, C. Lopez de Laorden, M. Menzel and G. Rajaraman, Angew. Chem., Int. Ed., 2007, 46, 8067 CrossRef CAS PubMed
- C. C. Reddy, Biological Oxidation Systems, Academic Press, 1990, vol. 1, pp. 297–327 Search PubMed
-
(a) P. Comba, M. Merz and H. Pritzkow, Eur. J. Inorg. Chem., 2003, 1711 CrossRef CAS PubMed
- K. Born, P. Comba, A. Daubinet, A. Fuchs and H. Wadepohl, J. Biol. Inorg. Chem., 2007, 12, 36 CrossRef CAS PubMed
-
(a) W. Susanto, C.-Y. Chu, W. J. Ang, T.-C. Chou, L.-C. Lo and Y. Lam, Green Chem., 2012, 14, 77 RSC
-
(a) W. Partenheimer, Adv. Synth. Catal., 2006, 348, 559 CrossRef CAS PubMed
-
(a) Ullman’s Encyclopaedia of Industrial Chemistry, 6th edn, Wiley-VCH, Weinheim, 2002 Search PubMed
-
(a) J. M. Hoover and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 16901 CrossRef CAS PubMed
- P. C. B. Page and T. McCarthy, Comprehensive Organic Synthesis, Pergamon, Oxford, UK, 1991, vol. 7, pp. 83–149 Search PubMed
-
(a) M. A. Fousteric, A. I. Koutsourea, S. S. Nikoloaropoulos, A. Riahi and J. Muzart, J. Mol. Catal., 2006, 250, 70 CrossRef PubMed
-
(a) Y. Miyahara, K. Goto and T. Inazu, Tetrahedron Lett., 2001, 42, 3097 CrossRef CAS
-
(a) P. S. Gradeff, US Patent 3, 840, 601, 1974
-
(a) Y. Peng and G. Song, Indian J. Chem., 2003, 42B, 924 CAS
-
(a) C. Chen, H. Yang, J. Chen, R. Zhang, L. Guo, H. Gan, B. Song, W. Zhu, L. Hua and Z. Hou, Catal. Commun., 2014, 47, 49 CrossRef CAS PubMed
- P. Comba, M. Kerscher and W. Schiek, Prog. Inorg. Chem., 2007, 55, 613 CrossRef CAS PubMed
- M. Haberberger, C. I. Someya, A. Company, E. Irran and S. Enthaler, Catal. Lett., 2012, 142, 557 CrossRef CAS
- P. Comba, H. Rudolf and H. Wadepohl, Dalton Trans., 2015, 44, 2724 RSC
- D. S. C. Black, G. B. Deacon and M. Rose, Tetrahedron, 1995, 51, 2055 CrossRef CAS
- K. Moriyama, M. Takemura and H. Togo, Org. Lett., 2012, 14, 2414 CrossRef CAS PubMed
-
(a) A. J. Fatiadi, Synthesis, 1978, 165 CrossRef CAS
-
(a) S. P. Rose and R. Smith, New Sci., 1969, 43, 468 CAS
-
(a) C. Ratan, United States Patent: 0059258, 2007
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C spectra of compounds L1, L3, FL1–FL3, p2′, 2, 7a–w, 9a–d, 11a–l, 13a–x. See DOI: 10.1039/c5ra17093a |
| This journal is © The Royal Society of Chemistry 2015 |