
Experimental and computational evidence for KOt-Bu-promoted synthesis of oxopyrazino[1,2-a]indoles†
Mohammad Mahdavia,
Rasoul Hassanzadeh-Soureshjanb,
Mina Saeedicd,
Alireza Ariafard*ef,
Rasool BabaAhmadif,
Parviz Rashidi Ranjbarb and
Abbas Shafiee*a
aDepartment of Medicinal Chemistry, Faculty of Pharmacy and Pharmaceutical Sciences Research Center, Tehran University of Medical Sciences, Tehran, Iran. E-mail: ashafiee@ams.ac.ir
bSchool of Chemistry, University College of Science, University of Tehran, PO Box 14155-6455, Tehran, Iran
cMedicinal Plants Research Center, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran
dPersian Medicine and Pharmacy Research Center, Tehran University of Medical Sciences, Tehran, Iran
eSchool of Physical Sciences (Chemistry), University of Tasmania, Private Bag 75, Hobart TAS 7001, Australia. E-mail: alirezaa@utas.edu.au
fDepartment of Chemistry, Faculty of Science, Central Tehran Branch, Islamic Azad University, Shahrak Gharb, Tehran, Iran
First published on 18th November 2015
Abstract
A novel series of oxopyrazino[1,2-a]indole derivatives were prepared via a two-step synthetic procedure including a Ugi-four-component reaction followed by the transition metal-free intramolecular hydroamination of Ugi adducts in the presence of KOt-Bu in DMF at room temperature. Density functional theory (DFT) calculations were also performed to elucidate the mechanistic aspects of the reaction.
1. Introduction
Natural and non-natural N-heterocyclic compounds constitute the most important part of biologically active structures having crucial roles in medicinal chemistry. Among the vast heterocyclic structural range, indole moieties have occupied a position of major importance and are known as anti-inflammatory,1 antibacterial,2 antidepressants,3 and analgesics.4 Structurally remarkable alkaloids possessing significant biological properties5 such as anti-tumor,6 anti-depression,7 cytotoxic and anti-microbial8 activities consist of an indole skeleton. At this juncture, fused indole derivatives have attracted significant interest over the past decades owing to a wide variety of pharmacological properties.9 Among various fused indole, pyrazino[1,2-a]indole scaffold absorbed our attention since they act as melatoninergic ligands,10 histamine H3 receptor,11 inhibitors of geranylgeranyltransferase I,12 selective imidazoline I2 receptor ligands,13 5-HT2C and dual 5-HT2C/5-HT6 receptor agonists.14,15 Also, they have depicted antifungal,16 antiproliferative,17 antibacterial,18 and anticancer19 activities. In spite of the efficacy of pyrazino[1,2-a]indoles, literature lacks adequate reports for their synthesis.20–24 Hence, development of novel, efficient, easy, and rapid synthetic approaches is still a common research area. Intramolecular cyclization of several 2-carbonyl-1-propargylindoles in the presence of ammonia was developed by Abbiati et al.20 The reaction was investigated in various heating conditions in the presence of different metal catalysts. Also, in the study reported by Laliberté et al.21 pyrazino[1,2-a]indole derivatives were prepared through palladium-catalyzed double allylic alkylation of indole-2-hydroxamates. Katritzky et al.24 studied three-step synthesis of 10-methyl-1,2,3,4-tetrahydropyrazino-[1,2-a]indoles starting from 2-(3-methyl-1H-indol-1-yl)ethylamine. It reacted with 2-chloroethan-1-amine, and then benzotriazole/formaldehyde. Finally, the corresponding derivatives were obtained via various nucleophilic substitutions using allylsilanes, silyl enol ether, and Grignard reagents.The intramolecular and intermolecular addition of an amine to an unsaturated carbon–carbon bond known as hydroamination reaction, has been powerful and versatile tool for the direct formation of C–N bond affording N-containing heterocycles.25–27 Comprehensive literature review related to hydroamination reactions showed that they usually need to be catalyzed by metals.25–27 Consequently, developing an efficient protocol in the absence of complex and transition metal-free catalysts would be worthwhile from the synthetic organic point of view.
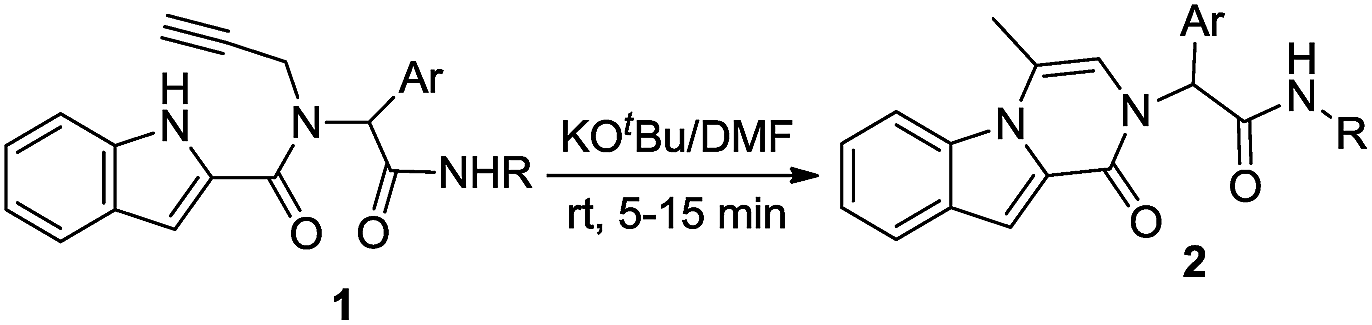
In view of our interest in the synthesis of novel heterocyclic compounds28 using isocyanide-based multicomponent reactions (IMCRs) as well as hydroamination reaction, we have developed an efficient, simple, and rapid synthesis of oxopyrazino[1,2-a]indole derivatives 2 (Scheme 1).
 | ||
| Scheme 1 Synthesis of novel oxopyrazino[1,2-a]indoles 2. | ||
2. Experimental
2.1. Reagents
Melting points were taken on a Kofler hot stage apparatus and are uncorrected. 1H and 13C NMR spectra were recorded on Bruker FT-500 and 400 using TMS as an internal standard. The IR spectra were obtained on a Nicolet Magna FTIR 550 spectrophotometer (in KBr). Mass spectra were documented on an Agilent Technology (HP) mass spectrometer operating at an ionization potential of 70 eV. The elemental analysis was performed on an Elementar Analysensystem. GmbH Vario EL CHNS mode.2.2. Synthesis of Ugi adduct 1; general procedure
A mixture of indole-2-carboxylic acid 3 (1 mmol), aromatic aldehyde 4 (1 mmol), propargylamine 5 (1 mmol), and isocyanide 6 (1.2 mmol) were dissolved in methanol (10 mL) and stirred at room temperature for 8 h. After completion of reaction, the precipitated Ugi product 1 was filtered off, washed with aqueous methanolic solution (20%), dried, and used for further reactions.![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 3.86–3.90 (m, 1H, NCH), 4.38–4.54 (m, 2H, CH2), 6.12–6.13 (m, 2H, CH, H3), 7.13 (td, J = 8.0, 1.0 Hz, 1H, H6), 7.29 (td, J = 8.0, 1.0 Hz, 1H, H5), 7.38–7.44 (m, 7H, Ph, H4, H7), 7.67 (d, J = 8.0 Hz, 1H, NH), 9.48 (s, 1H, NH). 13C NMR (125 MHz, CDCl3): δC = 24.8, 25.5, 32.8, 38.2, 48.8, 64.8, 72.8, 79.3, 106.6, 111.7, 120.7, 122.4, 125.1, 127.1, 128.8, 128.9, 129.8, 130.3, 134.3, 136.0, 163.9, 168.3. Anal. calcd for C26H27N3O2: C, 75.52; H, 6.58; N, 10.16; found: C, 75.41; H, 6.71; N, 10.29.CH), 2.36 (s, 3H, CH3), 3.87–3.90 (m, 1H, NCH), 4.30–4.54 (m, 2H, CH2), 6.14–6.17 (m, 2H, CH, H3), 7.13 (t, J = 8.0 Hz, 1H, H6), 7.19 (d, J = 8.0 Hz, 2H, H3′, H5′), 7.22–7.32 (m, 3H, H4, H5, H7), 7.40 (d, J = 8.0 Hz, 2H, H2′, H6′), 7.67 (d, J = 8.0 Hz, 1H, NH), 9.52 (s, 1H, NH). 13C NMR (125 MHz, CDCl3): δC = 21.1, 24.8, 25.5, 32.8, 38.2, 48.7, 64.1, 72.8, 80.0, 106.5, 111.7, 120.6, 122.4, 125.0, 127.8, 128.6, 129.6, 129.7, 131.2, 136.0, 138.7, 163.9, 168.5. Anal. calcd for C27H29N3O2: C, 75.85; H, 6.48; N, 9.83; found: C, 76.04; H, 6.62; N, 9.98.CH), 3.82 (s, 3H, OCH3), 3.84–3.89 (m, 1H, NCH), 4.36–4.50 (m, 2H, CH2), 6.08–6.11 (m, 2H, CH, H3), 6.91 (d, J = 8.0 Hz, 2H, H3′, H5′), 7.13 (t, J = 8.0 Hz, 1H, H6), 7.30 (td, J = 8.0, 1.0 Hz, 1H, H5), 7.437 (m, 3H, H4, H2′, H6′), 7.41 (d, J = 8.0 Hz, 1H, H7), 7.67 (d, J = 8.0 Hz, 1H, NH), 9.35 (s, 1H, NH). 13C NMR (125 MHz, CDCl3): δC = 24.8, 25.5, 32.9, 38.2, 48.7, 55.3, 62.1, 72.7, 80.0, 106.5, 111.7, 114.3, 120.7, 122.5, 125.0, 126.2, 127.9, 128.1, 131.3, 136.0, 160.0, 163.9, 168.6. Anal. calcd for C27H29N3O3: C, 73.11; H, 6.59; N, 9.47; found: C, 73.28; H, 6.78; N, 9.67.CH), 3.89–3.92 (m, 1H, NCH), 4.52–4.56 (m, 2H, CH2), 6.00 (s, 1H, CH), 6.46 (s, 1H, H3), 7.13 (td, J = 8.0, 1.0 Hz, 1H, H6), 7.28 (td, J = 8.0, 1.0 Hz, 1H, H5), 7.33–7.35 (m, 3H, H4′, H5′, H6′), 7.40–7.43 (m, 2H, H3, H4), 7.56 (d, J = 7.5 Hz, 1H, H3′), 7.67 (d, J = 8.0 Hz, 1H, NH), 9.39 (s, 1H, NH). 13C NMR (125 MHz, CDCl3): δC = 24.7, 24.8, 25.4, 32.8, 32.9, 38.2, 48.9, 61.5, 72.3, 79.3, 106.4, 111.7, 120.7, 122.5, 125.0, 127.2, 127.8, 128.4, 129.8, 130.0, 130.4, 131.0, 132.4, 136.0, 163.1, 168.1. Anal. calcd for C26H26ClN3O2: C, 69.71; H, 5.85; N, 9.38; found: C, 69.84; H, 5.68; N, 9.21.CH), 2.95 (s, 6H, 2 × CH3), 3.88–3.91 (m, 1H, NCH), 4.38–4.46 (m, 2H, CH2), 6.00–6.07 (m, 2H, CH, H3), 6.70 (d, J = 9.0 Hz, 2H, H3′, H5′), 7.13 (td, J = 8.0, 1.0 Hz, 1H, H6), 7.26–7.31 (m, 4H, H4, H5, H2′, H6′), 7.41 (d, J = 8.0 Hz, 1H, H7), 7.67 (d, J = 8.0 Hz, 1H, NH), 9.25 (s, 1H, NH). 13C NMR (125 MHz, CDCl3): δC = 24.7, 24.8, 25.5, 32.9, 33.0, 38.1, 40.3, 48.7, 63.0, 72.4, 80.3, 106.4, 111.6, 112.4, 120.6, 121.5, 122.5, 124.0, 124.9, 127.8, 129.0, 130.9, 150.0, 163.2, 169.0. Anal. calcd for C28H32N4O2: C, 73.66; H, 7.06; N, 12.27; found: C, 73.82; H, 7.22; N, 12.41.CH), 2.37 (s, 3H, CH3), 4.48–4.53 (m, 2H, CH2), 5.95–6.03 (m, 2H, CH, H3), 7.13 (td, J = 8.0, 1.0 Hz, 1H, H6), 7.20 (d, J = 8.0 Hz, 2H, H3′, H5′), 7.28–7.37 (m, 4H, H4, H5, H2′, H6′), 7.42 (d, J = 8.0 Hz, 1H, H7), 7.67 (d, J = 8.0 Hz, 1H, NH), 9.37 (s, 1H, NH). 13C NMR (125 MHz, CDCl3): δC = 21.2, 28.7, 38.1, 52.0, 65.1, 72.6, 80.0, 106.5, 111.6, 120.6, 122.5, 125.0, 127.9, 128.6, 129.6, 129.8, 131.3, 135.9, 138.7, 163.8, 168.7. Anal. calcd for C25H27N3O2: C, 74.79; H, 6.78; N, 10.47; found: C, 74.88; H, 6.60; N, 10.38.CH), 3.82 (s, 3H, OCH3), 4.38–4.52 (m, 2H, CH2), 5.95–6.04 (m, 2H, CH, H3), 6.92 (d, J = 8.0 Hz, 2H, H3′, H5′), 7.13 (t, J = 8.0 Hz, 1H, H6), 7.27–7.30 (m, 2H, H4, H5), 7.37 (d, J = 8.0 Hz, 2H, H2′, H6′), 7.42 (d, J = 8.0 Hz, 1H, H7), 7.67 (d, J = 8.0 Hz, 1H, NH), 9.45 (s, 1H, NH). 13C NMR (125 MHz, CDCl3): δC = 28.6, 38.2, 51.9, 55.3, 65.1, 72.6, 80.1, 106.4, 111.7, 114.3, 120.6, 122.4, 125.0, 126.4, 127.8, 131.3, 135.8, 136.0, 160.0, 163.8, 168.8. Anal. calcd for C25H27N3O3: C, 71.92; H, 6.52; N, 10.06; found: C, 71.83; H, 6.41; N, 10.27.CH), 4.49–4.52 (m, 2H, CH2), 5.90 (s, 1H, CH), 6.32 (s, 1H, H3), 7.08 (t, J = 9.0 Hz, 1H, H5′), 7.14 (t, J = 8.0 Hz, 1H, H6), 7.20–7.23 (m, 2H, H4′, H6′), 7.30 (t, J = 8.0 Hz, 1H, H5), 7.38–7.43 (m, 2H, H4, H7), 7.53–7.55 (m, 1H, H3′), 7.67 (d, J = 8.0 Hz, 1H, NH), 9.32 (s, 1H, NH). 13C NMR (125 MHz, CDCl3): δC = 28.6, 38.2, 52.1, 65.1, 72.4, 79.5, 106.4, 111.7, 115.7 (d, JC–F = 21.2 Hz), 120.7, 122.1 (d, JC–F = 12.5 Hz), 122.4, 124.5, 125.0, 128.4, 128.5, 131.0, 136.0, 136.5, 162.1 (d, JC–F = 245.0 Hz), 164.0, 168.0. Anal. calcd for C24H24FN3O2: C, 71.09; H, 5.97; N, 10.36; found: C, 71.18; H, 6.22; N, 10.50.
CH), 3.86–3.90 (m, 1H, NCH), 4.38–4.54 (m, 2H, CH2), 6.12–6.13 (m, 2H, CH, H3), 7.13 (td, J = 8.0, 1.0 Hz, 1H, H6), 7.29 (td, J = 8.0, 1.0 Hz, 1H, H5), 7.38–7.44 (m, 7H, Ph, H4, H7), 7.67 (d, J = 8.0 Hz, 1H, NH), 9.48 (s, 1H, NH). 13C NMR (125 MHz, CDCl3): δC = 24.8, 25.5, 32.8, 38.2, 48.8, 64.8, 72.8, 79.3, 106.6, 111.7, 120.7, 122.4, 125.1, 127.1, 128.8, 128.9, 129.8, 130.3, 134.3, 136.0, 163.9, 168.3. Anal. calcd for C26H27N3O2: C, 75.52; H, 6.58; N, 10.16; found: C, 75.41; H, 6.71; N, 10.29.CH), 2.36 (s, 3H, CH3), 3.87–3.90 (m, 1H, NCH), 4.30–4.54 (m, 2H, CH2), 6.14–6.17 (m, 2H, CH, H3), 7.13 (t, J = 8.0 Hz, 1H, H6), 7.19 (d, J = 8.0 Hz, 2H, H3′, H5′), 7.22–7.32 (m, 3H, H4, H5, H7), 7.40 (d, J = 8.0 Hz, 2H, H2′, H6′), 7.67 (d, J = 8.0 Hz, 1H, NH), 9.52 (s, 1H, NH). 13C NMR (125 MHz, CDCl3): δC = 21.1, 24.8, 25.5, 32.8, 38.2, 48.7, 64.1, 72.8, 80.0, 106.5, 111.7, 120.6, 122.4, 125.0, 127.8, 128.6, 129.6, 129.7, 131.2, 136.0, 138.7, 163.9, 168.5. Anal. calcd for C27H29N3O2: C, 75.85; H, 6.48; N, 9.83; found: C, 76.04; H, 6.62; N, 9.98.CH), 3.82 (s, 3H, OCH3), 3.84–3.89 (m, 1H, NCH), 4.36–4.50 (m, 2H, CH2), 6.08–6.11 (m, 2H, CH, H3), 6.91 (d, J = 8.0 Hz, 2H, H3′, H5′), 7.13 (t, J = 8.0 Hz, 1H, H6), 7.30 (td, J = 8.0, 1.0 Hz, 1H, H5), 7.437 (m, 3H, H4, H2′, H6′), 7.41 (d, J = 8.0 Hz, 1H, H7), 7.67 (d, J = 8.0 Hz, 1H, NH), 9.35 (s, 1H, NH). 13C NMR (125 MHz, CDCl3): δC = 24.8, 25.5, 32.9, 38.2, 48.7, 55.3, 62.1, 72.7, 80.0, 106.5, 111.7, 114.3, 120.7, 122.5, 125.0, 126.2, 127.9, 128.1, 131.3, 136.0, 160.0, 163.9, 168.6. Anal. calcd for C27H29N3O3: C, 73.11; H, 6.59; N, 9.47; found: C, 73.28; H, 6.78; N, 9.67.CH), 3.89–3.92 (m, 1H, NCH), 4.52–4.56 (m, 2H, CH2), 6.00 (s, 1H, CH), 6.46 (s, 1H, H3), 7.13 (td, J = 8.0, 1.0 Hz, 1H, H6), 7.28 (td, J = 8.0, 1.0 Hz, 1H, H5), 7.33–7.35 (m, 3H, H4′, H5′, H6′), 7.40–7.43 (m, 2H, H3, H4), 7.56 (d, J = 7.5 Hz, 1H, H3′), 7.67 (d, J = 8.0 Hz, 1H, NH), 9.39 (s, 1H, NH). 13C NMR (125 MHz, CDCl3): δC = 24.7, 24.8, 25.4, 32.8, 32.9, 38.2, 48.9, 61.5, 72.3, 79.3, 106.4, 111.7, 120.7, 122.5, 125.0, 127.2, 127.8, 128.4, 129.8, 130.0, 130.4, 131.0, 132.4, 136.0, 163.1, 168.1. Anal. calcd for C26H26ClN3O2: C, 69.71; H, 5.85; N, 9.38; found: C, 69.84; H, 5.68; N, 9.21.CH), 2.95 (s, 6H, 2 × CH3), 3.88–3.91 (m, 1H, NCH), 4.38–4.46 (m, 2H, CH2), 6.00–6.07 (m, 2H, CH, H3), 6.70 (d, J = 9.0 Hz, 2H, H3′, H5′), 7.13 (td, J = 8.0, 1.0 Hz, 1H, H6), 7.26–7.31 (m, 4H, H4, H5, H2′, H6′), 7.41 (d, J = 8.0 Hz, 1H, H7), 7.67 (d, J = 8.0 Hz, 1H, NH), 9.25 (s, 1H, NH). 13C NMR (125 MHz, CDCl3): δC = 24.7, 24.8, 25.5, 32.9, 33.0, 38.1, 40.3, 48.7, 63.0, 72.4, 80.3, 106.4, 111.6, 112.4, 120.6, 121.5, 122.5, 124.0, 124.9, 127.8, 129.0, 130.9, 150.0, 163.2, 169.0. Anal. calcd for C28H32N4O2: C, 73.66; H, 7.06; N, 12.27; found: C, 73.82; H, 7.22; N, 12.41.CH), 2.37 (s, 3H, CH3), 4.48–4.53 (m, 2H, CH2), 5.95–6.03 (m, 2H, CH, H3), 7.13 (td, J = 8.0, 1.0 Hz, 1H, H6), 7.20 (d, J = 8.0 Hz, 2H, H3′, H5′), 7.28–7.37 (m, 4H, H4, H5, H2′, H6′), 7.42 (d, J = 8.0 Hz, 1H, H7), 7.67 (d, J = 8.0 Hz, 1H, NH), 9.37 (s, 1H, NH). 13C NMR (125 MHz, CDCl3): δC = 21.2, 28.7, 38.1, 52.0, 65.1, 72.6, 80.0, 106.5, 111.6, 120.6, 122.5, 125.0, 127.9, 128.6, 129.6, 129.8, 131.3, 135.9, 138.7, 163.8, 168.7. Anal. calcd for C25H27N3O2: C, 74.79; H, 6.78; N, 10.47; found: C, 74.88; H, 6.60; N, 10.38.CH), 3.82 (s, 3H, OCH3), 4.38–4.52 (m, 2H, CH2), 5.95–6.04 (m, 2H, CH, H3), 6.92 (d, J = 8.0 Hz, 2H, H3′, H5′), 7.13 (t, J = 8.0 Hz, 1H, H6), 7.27–7.30 (m, 2H, H4, H5), 7.37 (d, J = 8.0 Hz, 2H, H2′, H6′), 7.42 (d, J = 8.0 Hz, 1H, H7), 7.67 (d, J = 8.0 Hz, 1H, NH), 9.45 (s, 1H, NH). 13C NMR (125 MHz, CDCl3): δC = 28.6, 38.2, 51.9, 55.3, 65.1, 72.6, 80.1, 106.4, 111.7, 114.3, 120.6, 122.4, 125.0, 126.4, 127.8, 131.3, 135.8, 136.0, 160.0, 163.8, 168.8. Anal. calcd for C25H27N3O3: C, 71.92; H, 6.52; N, 10.06; found: C, 71.83; H, 6.41; N, 10.27.CH), 4.49–4.52 (m, 2H, CH2), 5.90 (s, 1H, CH), 6.32 (s, 1H, H3), 7.08 (t, J = 9.0 Hz, 1H, H5′), 7.14 (t, J = 8.0 Hz, 1H, H6), 7.20–7.23 (m, 2H, H4′, H6′), 7.30 (t, J = 8.0 Hz, 1H, H5), 7.38–7.43 (m, 2H, H4, H7), 7.53–7.55 (m, 1H, H3′), 7.67 (d, J = 8.0 Hz, 1H, NH), 9.32 (s, 1H, NH). 13C NMR (125 MHz, CDCl3): δC = 28.6, 38.2, 52.1, 65.1, 72.4, 79.5, 106.4, 111.7, 115.7 (d, JC–F = 21.2 Hz), 120.7, 122.1 (d, JC–F = 12.5 Hz), 122.4, 124.5, 125.0, 128.4, 128.5, 131.0, 136.0, 136.5, 162.1 (d, JC–F = 245.0 Hz), 164.0, 168.0. Anal. calcd for C24H24FN3O2: C, 71.09; H, 5.97; N, 10.36; found: C, 71.18; H, 6.22; N, 10.50.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 875, 1651 cm−1. 1H NMR (500 MHz, CDCl3): δH = 1.41 (s, 9H, tert-butyl), 2.15 (s, 1H, CH), 4.49–4.53 (m, 2H, CH2), 5.85 (s, 1H, CH), 6.35 (s, 1H, H3), 7.13 (t, J = 8.0 Hz, 1H, H6), 7.29 (t, J = 8.0 Hz, 1H, H5), 7.20–7.23 (m, 3H, H4′, H5′, H6′), 7.32–7.43 (m, 2H, H4, H7), 7.59 (d, J = 7.5 Hz, 1H, H3′), 7.67 (d, J = 8.0 Hz, 1H, NH), 9.25 (s, 1H, NH). 13C NMR (125 MHz, CDCl3): δC = 28.7, 38.1, 52.0, 65.1, 72.2, 80.0, 106.4, 111.6, 120.7, 122.5, 125.0, 127.3, 128.0, 130.0, 130.3, 130.9, 131.4, 132.5, 136.1, 136.4, 163.1, 168.5. Anal. calcd for C24H24ClN3O2: C, 68.32; H, 5.73; N, 9.96; found: C, 68.51; H, 5.60; N, 10.17.
875, 1651 cm−1. 1H NMR (500 MHz, CDCl3): δH = 1.41 (s, 9H, tert-butyl), 2.15 (s, 1H, CH), 4.49–4.53 (m, 2H, CH2), 5.85 (s, 1H, CH), 6.35 (s, 1H, H3), 7.13 (t, J = 8.0 Hz, 1H, H6), 7.29 (t, J = 8.0 Hz, 1H, H5), 7.20–7.23 (m, 3H, H4′, H5′, H6′), 7.32–7.43 (m, 2H, H4, H7), 7.59 (d, J = 7.5 Hz, 1H, H3′), 7.67 (d, J = 8.0 Hz, 1H, NH), 9.25 (s, 1H, NH). 13C NMR (125 MHz, CDCl3): δC = 28.7, 38.1, 52.0, 65.1, 72.2, 80.0, 106.4, 111.6, 120.7, 122.5, 125.0, 127.3, 128.0, 130.0, 130.3, 130.9, 131.4, 132.5, 136.1, 136.4, 163.1, 168.5. Anal. calcd for C24H24ClN3O2: C, 68.32; H, 5.73; N, 9.96; found: C, 68.51; H, 5.60; N, 10.17.2.3. Synthesis of oxopyrazino[1,2-a]indole derivatives 2; general procedure
A mixture of Ugi product 1 (1 mmol) and potassium tert-butoxide (0.5 mmol) in dry DMF (7 mL) was stirred at room temperature for 5–15 min. After completion of reaction (checked by TLC), water (20 mL) was added to the reaction mixture and it was extracted with ethyl acetate (3 × 20 mL). The organic phase was dried over Na2SO4, the solvent was removed under reduced pressure, and the resulting residue was purified by column chromatography using ethyl acetate/petroleum ether = 1/3 as eluent and silica gel as stationary phase.2.4. Computational study
Gaussian 0931 was used to fully optimize all the structures involved in this study at the B3LYP level of density functional theory (DFT)32 in DMF using the CPCM solvation model.33 The 6-31G(d) basis set was used for all atoms.34 Frequency analyses were performed at the same level of theory to ensure that a minimum or transition state was achieved. Transition structures were located using the Berny algorithm and intrinsic reaction coordinate (IRC)35 calculations were used to confirm the connectivity between transition structures and minima. To further refine the energies obtained from the B3LYP/6-31G(d) calculations, we carried out single-point energy calculations for all of the structures with 6-311+G(2d,p) basis set in DMF using the CPCM solvation model at the M06 level. We have used the Gibbs free energies obtained from the M06/6-311+G(2d,p)//B3LYP/6-31G(d) calculations in DMF throughout the paper.
3. Results and discussion
After the introduction of novel multicomponent reactions (MCR),29 the obtained products from isocyanide-based multicomponent reactions (IMCRs) attracted lots of attention as they could perform further condensation or cyclization reactions due to the presence of versatile functional groups.28a,30 In this study, we focused on the Ugi adducts 1 obtained by the simultaneous reaction between indole-2-carboxylic acid 3, aromatic aldehydes 4, propargylamine 5, and isocyanides 6 (Scheme 1).For the preparation of desirable starting materials 1, four-component reaction of indole-2-carboxylic acid (1 mmol) 3, various aromatic aldehydes 4 (1 mmol), propargylamine 5 (1 mmol), and isocyanides 6 (1.2 mmol) were conducted in methanol at room temperature in the absence of any catalysts or additives within 8 h (Scheme 1). Subsequently, the intramolecular hydroamination reaction of compound 1 leading to the formation of product 2 (Scheme 1) was comprehensively studied. For this purpose, N-(2-(cyclohexylamino)-2-oxo-1-phenylethyl)-N-(prop-2-yn-1-yl)-1H-indole-2-carboxamide (1a) was chosen as the test substrate. To obtain product 2a in a rapid and efficient method, the effect of various conditions such as temperature, different solvents, and bases were investigated (Table 1).
| Entry | Solvent | Reagenta | Temperature | Time | Yield b |
|---|---|---|---|---|---|
| a All reactions were performed in the presence of 0.5 mmol of reagent.b Isolated yield. | |||||
| 1 | DMF | KOt-Bu | rt | 10 min | 75 |
| 2 | DMF | KOt-Bu | 45 °C | 10 min | 75 |
| 3 | DMF | KOt-Bu | 90 °C | 10 min | 30 |
| 4 | DMSO | KOt-Bu | rt | 30 min | 65 |
| 5 | DMSO | KOt-Bu | 50 °C | 30 min | 65 |
| 6 | DMF | K2CO3 | rt | 2 h | 45 |
| 7 | DMF | NEt3 | rt | 2 h | 10 |
| 8 | DMF | NaOH | rt | 1 h | 50 |
| 9 | DMF | L-proline | rt | 1 h | 15 |
| 10 | THF | KOt-Bu | rt | 1 h | 60 |
| 11 | 1,4-Dioxane | KOt-Bu | rt | 1 h | 55 |
| 12 | Toluene | KOt-Bu | rt | 1 h | 40 |
| 13 | CH2Cl2 | KOt-Bu | rt | 1 h | 35 |
Recently, we have described synthesis of novel benzo[6,7][1,4]oxazepino[4,5-a]quinazolinones via 7-exo-dig hydroamination of 3-substituted-2-[2-(prop-2-yn-1-yloxy)phenyl]-2,3-dihydroquinazolin-4(1H)-ones in the presence of potassium tert-butoxide (KOt-Bu) in DMF at 130 °C.28d It was revealed that KOt-Bu/DMF system could efficiently promote the corresponding hydroamination. The efficiency of KOt-Bu was previously confirmed by Polindara-García et al. in the hydroamination reaction of some Ugi adducts.30b Those results led us to examine the aforementioned model reaction under similar condition as well as all conditions depicted in Table 1. As shown in Table 1, product 2a was obtained in the presence of KOt-Bu in DMF at room temperature (Table 1, entry 1), in very short reaction time and good yields. It was found that 0.5 equivalent amount of base was sufficient and higher amounts did not improve the yield of reaction. Also, we perceived that the model reaction was efficiently conducted at room temperature and higher temperature not only did not increase the yield of reaction but also decreased it (Table 1, entry 3).
With these results in hand, various oxopyrazino[1,2-a]indole derivatives 2a–i were synthesized using various Ugi adducts 1 (Table 2). All substrates possessing electron-rich as well as electron-poor substituents underwent intramolecular hydroamination reaction leading to the formation of the related products 2 in very short reaction time (5–15 min) and good yields (75–80%). All products were characterized using IR, NMR spectroscopy as well as mass spectrometry and all data confirmed the structures of the synthesized compounds.
|
||||
|---|---|---|---|---|
| Entry | Ar | R | Product 2 | Yielda |
| a Isolated yield. | ||||
| 1 | C6H5 | Cyclohexyl | 2a | 75 |
| 2 | 4-Me-C6H4 | Cyclohexyl | 2b | 77 |
| 3 | 4-MeO-C6H4 | Cyclohexyl | 2c | 75 |
| 4 | 2-Cl-C6H4 | Cyclohexyl | 2d | 70 |
| 5 | 4-N(Me)2-C6H4 | Cyclohexyl | 2e | 75 |
| 6 | 4-Me-C6H4 | tert-Butyl | 2f | 80 |
| 7 | 4-MeO-C6H4 | tert-Butyl | 2g | 77 |
| 8 | 2-F-C6H4 | tert-Butyl | 2h | 78 |
| 9 | 2-Cl-C6H4 | tert-Butyl | 2i | 75 |
As shown in Scheme 2, the intramolecular hydroamination reaction occurred through 6-exo-dig ring closure system followed by [1,3]-H shift to afford product 2. It was clear that no product was obtained through 5-endo-dig ring closure which is probable as reported by Polindara-García et al.30b In the mentioned report, Ugi adducts were prepared by the reaction of aldehydes, isocyanides, carboxylic acids, and propargylamine. Then, KOt-Bu-mediated ring closure afforded 2,3-dihydropyrroles through 5-endo cycloisomerization of the later adducts. It is clear that presence of indole NH in our study played important role to change the mode of ring closure.
 | ||
| Scheme 2 Reaction sequences for the construction of oxopyrazino[1,2-a]indoles 2. | ||
In continuation of our investigations to gain further understanding of probable mechanism of the reaction, density functional theory (DFT) calculations were performed. Two major pathways were investigated for the corresponding C–N bond formation catalyzed by KOt-Bu. In pathway A (Fig. 1a), the reaction starts with the deprotonation of the indole N–H proton using −Ot-Bu followed by nucleophilic attack of the nitrogen anion on the internal C2 alkyne carbon to give vinyl anion intermediate 4m. This intermediate is capable of re-deprotonating the alcohol formed in situ (HOt-Bu) to produce the alkene intermediate 5m. Eventually, proton abstraction from 5m by −Ot-Bu gives allyl anion intermediate 6m and the final product 2m is formed through a proton transfer from HOt-Bu to 6m. In pathway B (Fig. 1b), the C3 of 1m is deprotonated by −Ot-Bu to yield allenyl anion 7m. Internal proton transfer from the indole N–H to C1 atom of 7m followed by attack of the nitrogen anion on the C2 atom of 8m gives the allyl anion 6m from which product 2m is formed via the sequence 6m → TS6-2 → 2m (as discussed in pathway A). As the N–H deprotonation (the first step of pathway A) is calculated to proceed via a barrierless process,36 the reaction is expected to initiate on the pathway A; the C3 deprotonation with ΔG‡ = 5.3 kcal mol−1 (the first step of pathway B) is much less favorable than the indole N–H deprotonation. On the other hand, the reaction of 1m with −Ot-Bu on pathway A is predicted to be irreversible due to the fact that the energy of all transition structures on this pathway (TS3-4, TS4-5, TS5-6, and TS6-2) lies below the energy of 1m. All the transition structures of pathway A are calculated to be lower in energy than the vital transition structures of pathway B (TS1-7 and TS7-8). We have also investigated another alternative pathway for the 6-exo-dig ring closure process in which the alkyne terminal CH proton is deprotonated followed by the intramolecular deprotonation of NH indole group.37 Although the alkyne terminal CH proton is acidic and no transition structure was found for its deprotonation, this pathway is not favorable at all because transition structure relating to the intramolecular NH deprotonation with a relative Gibbs energy of 13.9 kcal mol−1 lies above all the calculated transition structures. From these results, it can be concluded that pathway A is most likely operative for formation of product 2.
 | ||
| Fig. 1 Potential energy profiles relating to formation of oxopyrazino[1,2-a]indoles 2m. The relative free energies obtained from the M06/6-311+G(2d,p)//B3LYP/6-31G(d) calculations in DMF are given in kcal mol−1. | ||
On the basis of the calculations, reaction of 1m → 2m is exergonic by −47.2 kcal mol−1 and the rate determining step for this reaction is computed to be attack of the nitrogen anion on the internal C2 alkyne carbon (transformation 3m → 4m) with an activation energy of 20.5 kcal mol−1. The detailed catalytic cycle of the −Ot-Bu-catalyzed intramolecular hydroamination reaction is summarized in Scheme 3. Striking features that emerge from the calculations are that, in this catalytic reaction, the −Ot-Bu catalyst is consumed and regenerated several times during the catalytic cycle (Scheme 3) and proton transfers are not likely to occur without the assistance of the conjugate acid–base pairs (−Ot-Bu/HOt-Bu). For example, the activation barrier for conversion of 4m to 6m in the absence of tBuO−/tBuOH is calculated to be as high as 41.5 kcal mol−1 while it reduces to 15.2 kcal mol−1 in the presence of the acid–base pairs.
 | ||
| Scheme 3 Intramolecular hydroamination reaction. | ||
We now turn our attention to address why the 5-endo-dig ring closure does not occur for these substrates (Scheme 2). The first step for this process is surmised to be deprotonation of the carbon bound to the phenyl group (Fig. 2). Our calculations show that the activation energy for the deprotonation via TS1-9 (10.0 kcal mol−1) is much higher in energy than that for the deprotonation of the indole N–H (Fig. 1a).
 | ||
| Fig. 2 Potential energy profile for the 5-endo-dig ring closure process. The relative free energies obtained from the M06/6-311+G(2d,p)//B3LYP/6-31G(d) calculations in DMF are given in kcal mol−1. | ||
The transformation 1m + tBuO− → 9m is about 12.0 kcal mol−1 less exergonic than the transformation 1m + tBuO− → 3m. These results suggest that the indole N–H proton is more acidic, resulting in more favorable 6-exo-dig ring closure. Indeed, the acidity of the proton being initially deprotonated mainly controls the chemoselectivity of the catalytic reaction.38
4. Conclusion
In conclusion, a two-step synthetic procedure for the synthesis of novel oxopyrazino[1,2-a]indole derivatives was described via a convenient Ugi 4-CR of indole-2-carboxylic acid, various aromatic aldehydes, propargylamine, and isocyanides, followed by the intramolecular hydroamination cyclization of indolic NH group and propargylic triple bond in the presence of KOt-Bu in DMF at room temperature. All products were obtained in very short reaction time (5–15 min) and good yields which make it a practical protocol for the library-based synthesis of oxopyrazino[1,2-a]indoles as well as investigation of miscellaneous biological activities. Also, density functional theory (DFT) calculations were applied to gain insight into the mechanism of reaction (Scheme 3). It seems that the catalytic reaction is started by the deprotonation of the indole N–H, followed by nucleophilic attack of the nitrogen anion on the internal C2 alkyne carbon to obtain a vinyl anion. This vinyl intermediate then deprotonates HOt-Bu to give an alkene intermediate. Finally, an allyl anion formed by deprotonation of the alkene intermediate abstracts a proton from HOt-Bu and gives the final product.Acknowledgements
This research was supported by grants from the Research Council of Tehran University of Medical Sciences and Iran National Science (INSF).References
- N. Singh, S. K. Bhati and A. Kumar, Eur. J. Med. Chem., 2008, 43, 2597 CrossRef CAS PubMed.
- J. D. Williams, S. T. Nguyen, S. Gu, X. Ding, M. M. Butler, T. F. Tashjian, T. J. Opperman, T. L. Bowlin, R. G. Panchal, S. Bavari, N. P. Peet and D. T. Moir, Bioorg. Med. Chem. Lett., 2013, 21, 7790 CrossRef CAS PubMed.
- D. Zhou, P. Zhou, D. A. Evrard, K. Meagher, M. Webb, B. L. Harrison, D. M. Huryn, J. Golembieski, G. A. Hornby, L. E. Schechter, D. L. Smith, T. H. Andree and R. E. Mewshaw, Bioorg. Med. Chem., 2008, 16, 6707 CrossRef CAS PubMed.
- H. Takayama, Chem. Pharm. Bull., 2004, 52, 916 CrossRef CAS PubMed.
- (a) S. E. O'Connor and J. J. Maresh, Nat. Prod. Rep., 2006, 23, 532 RSC; (b) A. Aygün and U. Pindur, Curr. Med. Chem., 2003, 10, 1113 CrossRef; (c) L. Gupta, A. Talwar and P. M. S. Chauhan, Curr. Med. Chem., 2007, 14, 1789 CrossRef CAS PubMed; (d) W. Gul and M. T. Hamann, Life Sci., 2005, 78, 442 CrossRef CAS PubMed.
- (a) H. Fujiki, M. Mori, M. Nakayasu, M. Terada, T. Sugimura and R. E. Moore, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 3872 CrossRef CAS PubMed; (b) K. Irie, N. Hagiwara, H. Tokuda and K. Koshimizu, Carcinogenesis, 1987, 8, 547 CAS.
- A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489 CrossRef CAS PubMed.
- S. K. Adla, F. Sasse, G. Kelter, H.-H. Fiebig and T. Lindel, Org. Biomol. Chem., 2013, 11, 6119 CAS.
- N. K. Kaushik, N. Kaushik, P. Attri, N. Kumar, C. H. Kim, A. K. Verma and E. H. Choi, Molecules, 2013, 18, 6620 CrossRef CAS PubMed.
- C. Markl, M. I. Attia, J. Julius, S. Sethi, P. A. Witt-Enderby and D. P. Zlotos, Bioorg. Med. Chem., 2009, 17, 4583 CrossRef CAS PubMed.
- H. G. Richter, C. Freichel, J. Huwyler, T. Nakagawa, M. Nettekoven, J. M. Plancher, S. Raab, O. Roche, F. Schuler, S. Taylor, C. Ullmer and R. Wiegand, Bioorg. Med. Chem. Lett., 2010, 20, 5713 CrossRef CAS PubMed.
- D. M. Vigushin, G. Brooke, D. Willows, R. C. Coombes and C. J. Moody, Bioorg. Med. Chem. Lett., 2003, 13, 3661 CrossRef CAS PubMed.
- J. Chang-Fong, R. J. Tyacke, A. Lau, J. Westaway, A. L. Hudson and R. A. Glennon, Bioorg. Med. Chem. Lett., 2004, 14, 1003 CrossRef CAS PubMed.
- S. Röver, D. R. Adams, A. Bénardeau, J. M. Bentley, M. J. Bickerdike, A. Bourson, I. A. Cliffe, P. Coassolo, J. E. Davidson, C. T. Dourish, P. Hebeisen, G. A. Kennett, A. R. Knight, C. S. Malcolm, P. Mattei, A. Misra, J. Mizrahi, M. Muller, R. H. Porter, H. Richter, S. Taylor and S. P. Vickers, Bioorg. Med. Chem. Lett., 2005, 15, 3604 CrossRef PubMed.
- N. Krogsgaard-Larsen, A. A. Jensen and J. Kehler, Bioorg. Med. Chem. Lett., 2010, 20, 5431 CrossRef CAS PubMed.
- R. K. Tiwari, A. K. Verma, A. K. Chhillar, D. Singh, J. Singh, V. K. Sankar, V. Yadav, G. L. Sharma and R. Chandra, Bioorg. Med. Chem., 2006, 14, 2747 CrossRef CAS PubMed.
- R. Romagnoli, P. G. Baraldi, M. D. Carrion, O. Cruz-Lopez, C. L. Cara, D. Preti, M. A. Tabrizi, J. Balzarini, E. Hamel, E. Fabbri and R. Gambari, Lett. Drug Des. Discovery, 2009, 6, 298 CrossRef CAS PubMed.
- R. K. Tiwari, D. Singh, J. Singh, J. V. Yadav, A. K. Pathak, R. Dabur, A. K. Chhillar, R. Singh, G. L. Sharma, R. Chandra and A. K. Verma, Bioorg. Med. Chem. Lett., 2006, 16, 413 CrossRef CAS PubMed.
- Z. Shiokawa, K. Hashimoto, B. Saito, Y. Oguro, H. Sumi, M. Yabuki, M. Yoshimatsu, Y. Kosugi, Y. Debori, N. Morishita, D. R. Dougan, G. P. Snell, S. Yoshida and T. Ishikawa, Bioorg. Med. Chem., 2013, 21, 7938 CrossRef CAS PubMed.
- (a) G. Abbiati, A. Arcadi, A. Bellinazzi, E. Beccalli, E. Rossi and S. Zanzola, J. Org. Chem., 2005, 70, 4088 CrossRef CAS PubMed; (b) E. M. Beccalli, A. Bernasconi, E. Borsini, G. Broggini, M. Rigamonti and G. Zecchi, J. Org. Chem., 2010, 75, 6923 CrossRef CAS PubMed.
- S. Laliberté, P. K. Dornan and A. Chen, Tetrahedron Lett., 2010, 51, 363 CrossRef.
- R. Toche, S. Chavan and R. Janrao, Monatsh. Chem., 2014, 145, 1507 CrossRef CAS.
- V. I. Shvedov, V. V. Alekseev, L. B. Altukhova and A. N. Grinev, Pharm. Chem. J., 1968, 2, 653 CrossRef.
- A. R. Katritzky, A. K. Verma, H.-Y. He and R. Chandra, J. Org. Chem., 2003, 68, 4938 CrossRef CAS PubMed.
- (a) T. E. Müller and M. Beller, Chem. Rev., 1998, 98, 675 CrossRef; (b) T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795 CrossRef PubMed; (c) F. Pohlki and S. Doye, Chem. Soc. Rev., 2003, 32, 104 RSC; (d) L. Llauger, C. Bergami, O. D. Kinzel, S. Lillini, G. Pescatore, C. Torrisi and P. Jones, Tetrahedron Lett., 2009, 50, 172 CrossRef CAS.
- H. Nagata, Y. Sugimoto, Y. Ito, M. Tanaka and M. Yoshimatsu, Tetrahedron, 2014, 70, 1306 CrossRef CAS.
- G.-Q. Liu, B. Cui, H. Sun and Y.-M. Li, Tetrahedron, 2014, 70, 5696 CrossRef CAS.
- (a) M. Saeedi, M. Mahdavi, A. Foroumadi and A. Shafiee, Tetrahedron, 2013, 69, 3506 CrossRef CAS; (b) M. Mahdavi, M. Asadi, M. Saeedi, M. Ebrahimi, M. A. Rasouli, P. R. Ranjbar, A. Foroumadi and A. Shafiee, Synthesis, 2012, 44, 3649 CrossRef CAS; (c) M. A. Rasouli, M. Mahdavi, P. R. Ranjbar, M. Saeedi, A. Shafiee and A. Foroumadi, Tetrahedron Lett., 2012, 53, 7088 CrossRef CAS; (d) M. Mahdavi, N. Foroughi, M. Saeedi, M. Karimi, H. Alinezhad, A. Foroumadi, A. Shafiee and T. Akbarzadeh, Synlett, 2014, 25, 385 CAS; (e) S. Nahavandian, S. Allameh, M. Saeedi, S. Ansari, M. Mahdavi, A. Foroumadi and A. Shafiee, Helv. Chim. Acta, 2015, 98, 1028 CrossRef CAS; (f) M. A. Rasouli, M. Mahdavi, M. Saeedi, P. R. Ranjbar, A. Shafiee and A. Foroumadi, J. Heterocycl. Chem., 2015, 52, 386 CrossRef CAS; (g) T. Akbarzadeh, S. Noushini, S. Taban, M. Mahdavi, M. Khoshneviszadeh, M. Saeedi, S. Emami, M. Eghtedari, Y. Sarrafi, M. Khoshneviszadeh, M. Safavi, K. Divsalar, M. H. Moshafi, A. Asadipour, R. Sabourian, N. Edraki, O. Firouzi, R. Miri, A. Shafiee and A. Foroumadi, Mol. Diversity, 2015, 19, 273 CrossRef CAS PubMed.
- (a) M. Passerini and L. Simone, Gazz. Chim. Ital., 1921, 51, 126 CAS; (b) I. Ugi, R. Meyr, U. Fetzer and C. Steinbrückner, Angew. Chem., 1959, 71, 386 Search PubMed.
- (a) A. Kumar, Z. Li, S. K. Sharma, V. S. Parmar and E. V. V. der Eycken, Org. Lett., 2013, 15, 1874 CrossRef CAS PubMed; (b) L. A. Polindara-García and L. D. Miranda, Org. Lett., 2012, 14, 5408 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- (a) C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS; (b) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161 CrossRef CAS; K. Fukui, Acc. Chem. Res., 1981, 14, 363 CrossRef.
- We are not able to locate any transition structure for this process due to the fact that the indole deprotonation has no enthalpic activation barrier, a result which is supported by a relaxed PES scan (Fig. S1†).
- Consider pathway C, Fig. S2.†.
- Although, in this system, the pKa values determine which mechanism is operative (Fig. S3†), it is not necessarily true to say that the reaction is always initiated from the most acidic proton. For example, if the transition structure TS3-4 was calculated to be higher in energy than transition structure TS1-7 (Fig. 1), then the reaction did proceed via pathway B and not pathway A. Therefore, in order to determine which pathway is responsible for producing the final product, we have to investigate all single steps of all possible pathways.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra17056g |
| This journal is © The Royal Society of Chemistry 2015 |